







Hello,hello,小伙伴们。到了秋高气爽的九月,不知道各位科研顺利么?今年的科研小目标达成了么?今天依旧为大家介绍单细胞及空间转录组相关内容。之前我们也给大家介绍过MERFISH等技术,今天我们详细的为大家介绍基于成像的单细胞转录组学和蛋白质组学定量方法。
今天我们为大家讲解STAMP技术,在《STAMP: Single-cell transcriptomics analysis and
multimodal profiling through imaging》这篇文章中,作者首先介绍了常规单细胞RNA测序的发展和局限等:
- scRNA-seq 的核心贡献:过去十年彻底改变对细胞多样性的认知,可提供单个细胞高分辨率图谱,助力解析复杂组织和器官在稳定状态及分化、疾病相关扰动等动态过程中的细胞组成与状态,还推动果蝇、小鼠、人类等全生物体细胞图谱构建。
- scRNA-seq 的成本问题:依赖测序且需对细胞单独索引,导致成本高昂,且成本随细胞数量呈线性增长。
- scRNA-seq 的转录本采样偏差:转录本随机采样会引入偏差,高丰度转录本过度呈现,转录因子等低表达基因被忽略。
- scRNA-seq 的技术效率缺陷:技术本身效率低,droplet-based 微流控技术易致细胞损伤、封装效率低和液滴不稳定,造成大量样本损失;基于平板的组合索引法存在细胞捕获有限、交叉污染和索引效率低的问题,且细胞大小、脆性、RNA 含量等因素会进一步影响捕获效率,导致部分细胞群体代表性不足或被遗漏,数据集无法准确反映样本真实细胞组成与复杂性。
- scRNA-seq 的通量与细胞破坏问题:通量较低,每次实验通常仅能处理数千个细胞,在超低(数百个细胞)和超高(数百万个细胞)规模分析上均存在挑战;测序过程会破坏细胞,无法将分子图谱与细胞结构、形态或功能属性相结合。
为解决 scRNA-seq 的上述问题,研究团队提出基于成像的单细胞转录组学和蛋白质组学读数方法,开发出 STAMP 技术,旨在实现大规模细胞分析、降低成本并保留单细胞特征检测优势,为单细胞研究领域带来新突破。
二、STAMP 技术核心原理
STAMP 技术创新性地将转录组学和蛋白质组学成像平台(如 Xenium Analyzer、CosMx Spatial Molecular Imager、MERSCOPE 和 PhenoCyler Fusion)改造为可扩展且灵活的单细胞分析工具。它借助成像技术替代传统测序,通过将悬浮细胞固定在载玻片上形成单层,实现对单细胞的多模态(RNA、蛋白质、H&E 染色)分析,在保留细胞结构和形态的同时,大幅降低成本并提高分析规模。
数据分析:运用多种生物信息学方法进行分析,包括无监督聚类(如 InSituType 算法)、主成分分析(PCA)、Harmony 整合去除批次效应、通路富集分析(如基于 MSigDB 数据库的 AUCell 分析)以及细胞轨迹分析(如 Palantir)等,以挖掘细胞类型、基因表达模式、细胞状态动态变化等信息。
三、主要研究结果
(一)无测序的单细胞基因组学分析
研究团队首先利用 CosMx SMI 平台(STAMP-C)和 1000-plex 人类通用细胞表征 RNA 面板对 LNCaP、MCF-7 和 SK-BR-3 三种癌细胞系进行分析。多样本载玻片阵列包含四个 sub-STAMP,每个 sub-STAMP 约有 35,000 个细胞,其中三个为单一癌细胞系,一个为 1:1:1 的混合细胞系。通过成像和严格的质量控制,平均每个细胞系获得 11,103 个细胞,每个细胞的中位数转录本数为 3,637,基因数为 413,仅 4.63% 的细胞被排除在分析之外。
使用 InSituType(IST)无监督聚类算法,在混合细胞系的 sub-STAMP 中识别出三个具有独特转录谱的不同聚类,每个视野(FOV)中每种细胞系的占比平均为 33.33%(范围:32.6%-33.4%)。将 IST 算法应用于单一细胞系的 sub-STAMP,也得到了三个不同的聚类,其转录谱与混合细胞系中的聚类相匹配。此外,混合细胞系和单一细胞系的基因特征与使用 Single-Cell Gene Expression Flex 实验(10× Genomics)对相同固定癌细胞悬浮液生成的 scRNA-seq 参考数据集高度相关,验证了 STAMP 技术在基于成像的转录组学分析中的适用性和特异性。
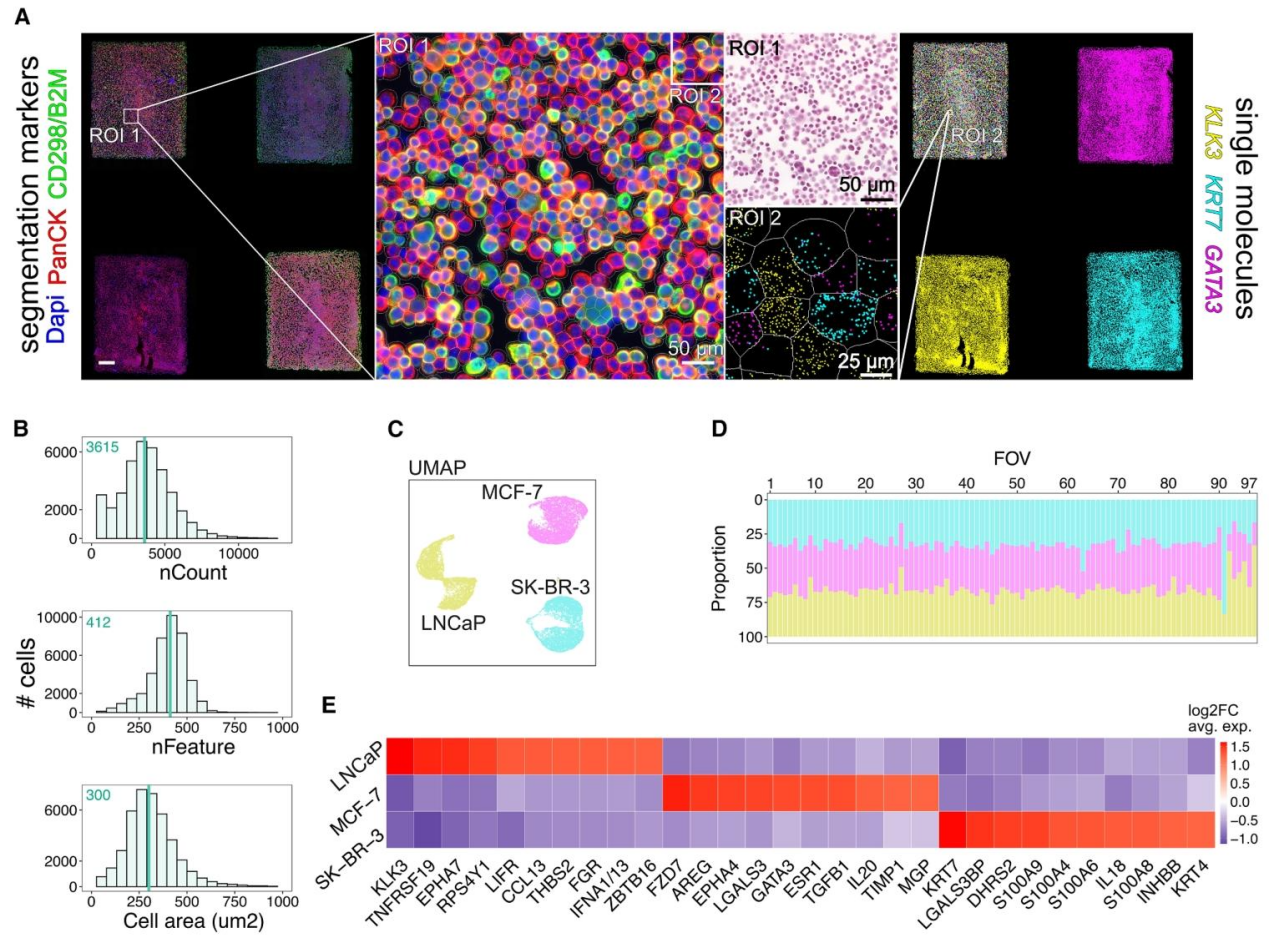
图 1. 基于成像的无测序单细胞基因组学分析
(二)低输入细胞数量的灵敏捕获
为探究 STAMP 技术在超低细胞密度下的可扩展性,研究人员将 MCF-7、SK-BR-3 和 LNCaP 三种癌细胞系按 1:1:1 的比例混合,制备成四个细胞数量不同(约 100、250、500 和 1,000 个细胞)的 sub-STAMP,同时还在两个 sub-STAMP 中分别分析了约 20,000 个细胞和 20,000 个细胞核,以直接比较高 RNA 含量(完整细胞)和低 RNA 含量(细胞核)样本的情况。
结果显示,高质量细胞的平均回收率为 85.33%(范围:65%-97%),每个 sub-STAMP 中每种细胞系的平均占比为 32.5%(范围:18.6%-56.9%)。在细胞数量较多的 sub-STAMP 中,每个细胞的中位数基因计数保持一致,而在细胞数量最少的 sub-STAMP(100 个细胞)中,检测到的基因数量有所减少。每个细胞检测到的特征数量也呈现类似趋势,表明在分析极低细胞密度时性能欠佳。在细胞密度较低的 sub-STAMP 中,细胞面积保持稳定,但在细胞密度较高的 sub-STAMP 中,细胞面积减少了一半,这可能是由于细胞分割过程中细胞扩张减少所致。此外,虽然细胞核的计数和特征数量少于完整细胞,但细胞和细胞核之间的基因表达水平高度相关。
为验证方法的可重复性,研究人员将 MCF-7 和 SK-BR-3 细胞悬浮液分成两份,在两个载玻片上作为技术重复进行成像(使用 Xenium Prime 5k 人类泛组织和通路面板,STAMP-X)。尽管回收的细胞数量略有不同,但两个重复样本在检测到的转录本和基因数量以及细胞面积方面均表现出高度一致性,且基因表达水平在重复样本之间高度相关。
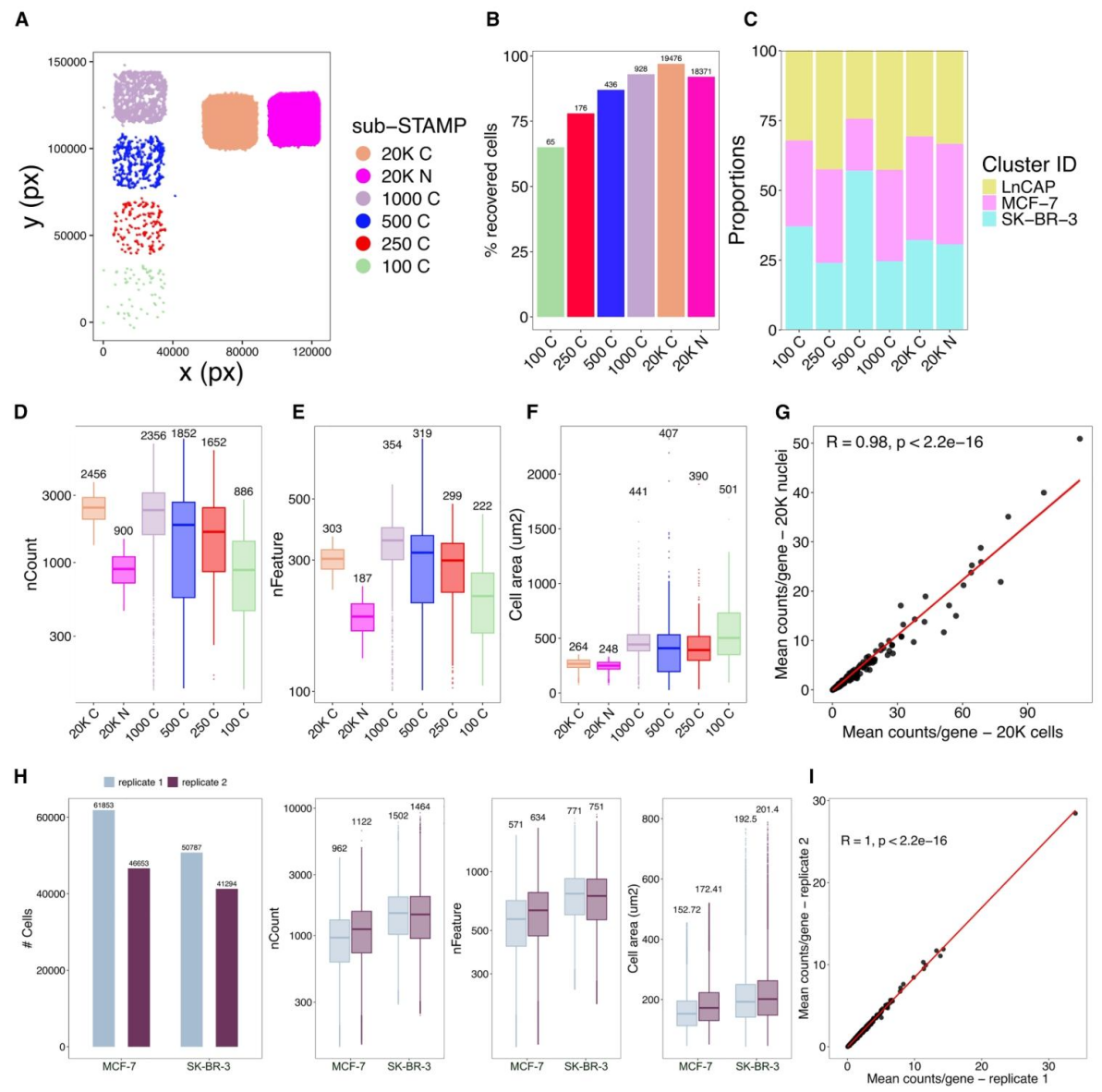
图 2. 对低输入细胞数量的灵敏捕获
(三)跨平台的样本多重分析
为评估 STAMP 技术的多重分析能力,研究人员制备了 27 个不同的样本,包括外周血单核细胞(PBMCs)、干细胞、癌细胞系、癌症相关成纤维细胞(CAFs)、从前列腺癌 FFPE 组织中提取的细胞核样本以及混合细胞群体。将这些样本分别压印在 STAMP-C 和 STAMP-X 两种实验的两个重复载玻片上,其中 21 个 sub-STAMP 在两种实验中共享,以确保一致性和可重复性,同时全面评估实验的多重分析能力。
在每个平台上,研究人员都选择了最大的基因面板以匹配 sub-STAMP 所代表的细胞类型多样性,即 Xenium Prime 5k 人类泛组织和通路面板以及 CosMx 人类 6K 发现面板。结果显示,每个细胞的基因和转录本数量以及细胞面积在重复样本之间高度相关,再次证明了实验的稳健性。此外,在基因、分子计数和面积分布的上下限处的实验条件在不同平台之间也保持一致。对所有样本和重复样本的基因进行汇总后发现,不仅在重复样本内部,而且在 21 个条件中的 18 个条件下,不同平台之间也存在高度相关性。CHP-134、SK-N-DZ 和 KELLY 细胞系的相关性欠佳,可能是由于两个面板之间的重叠率较低(29.6%)所致。在生物学相似的样本之间也检测到高度相关性,例如 iPSC 32F、hESC 和 MD3iPSC 聚在一起,LNCaP 和 V16D(一种源自 LNCaP 细胞的去势抵抗性前列腺癌细胞系)也聚在一起,单个癌细胞系 LNCaP、SK-BR-3 和 MCF-7 与其混合样本(MX1 和 MX2)相比也呈现出类似趋势。
由于 STAMP 技术在不同平台之间具有高度相关性,研究人员对所有数据集一起进行了主成分分析(PCA)。使用 Harmony 整合成功去除了由面板和平台引入的批次效应(由 PC1 捕获),这种整合还提高了局部逆辛普森指数(LISI 得分),反映了跨平台(技术层面)和重复批次(实验层面)的一致性。随后,研究人员使用 AUCell 对 sub-STAMP 上的从MSigDB 数据库(https://www.gsea-msigdb.org/gsea/index.jsp)中的精选通路进行了分析。例如,在PBMCs 中识别出 Biocarta 淋巴细胞通路的特定富集,且在重复样本之间保持一致。在癌细胞系中,Hallmark 糖酵解和缺氧通路以及 GOBP 乳酸代谢过程也同样富集,但在骨髓祖细胞和前列腺癌 FFPE 细胞核中得分较低,这与癌细胞在缺氧条件下由于糖酵解速率增加而产生更多乳酸的代谢适应以及体外培养的分化细胞系的代谢特征相符。
研究人员还将 STAMP 技术应用于 MERSCOPE 平台(Vizgen),使用 PanNeuro 细胞类型面板(500 个基因)对之前分析过的 12 个细胞系进行了分析(STAMP-M)。总共分析了 285,445 个细胞,每个细胞的中位数转录本数为 194(SD=318.7),独特基因数为 102(SD=70.05)。STAMP-M 分析得到的细胞面积大小与之前的 STAMP 实验相似,大多数细胞质量较高(251,199 个细胞,占 88%)。通过一对一的差异表达分析,确定了每个细胞系的特定基因标志物,证实了 STAMP-M 在细胞表型分析中的适用性。这些结果表明,STAMP 技术能够在单个载玻片上实现高水平的多重分析,便于在各种成像平台上同时分析多个样本。
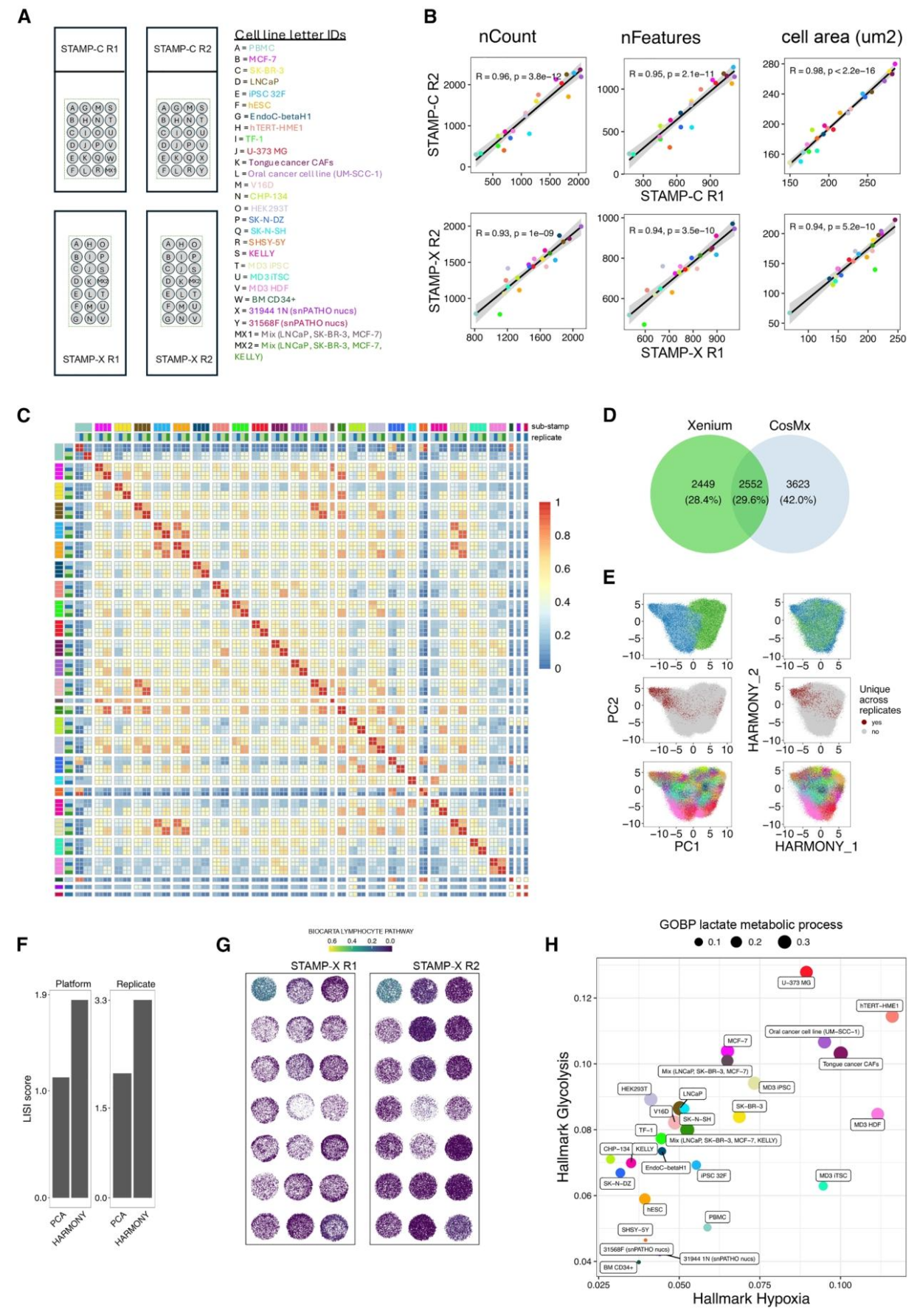
图 3. 跨平台的样本多重分析
(四)数百万循环血细胞的免疫表型分析
研究人员进一步扩展实验方案,在每次实验中对数百万个细胞进行STAMP分析。生成了一个包含约 170 万个 PBMCs 的高密度 STAMP,应用 Xenium 免疫肿瘤学面板(380 个目标基因)进行成像,每个细胞的中位数转录本数为 83(范围:27-259),基因数为 49(范围:24-103)。经过质量控制去除分割错误和计数及特征异常的细胞后,保留了 88.53% 的高质量细胞。随后进行常规的单细胞分析,包括降维和 PCA,结果显示主成分(PCs)1-3 能够根据细胞谱系标志物基因有效区分髓系、T 细胞和 B 细胞这三个主要免疫谱系,且髓系细胞群体显示出更多的基因和转录本数量以及更大的细胞面积。
对每个免疫谱系进行进一步聚类,根据细胞状态标志物识别出所有主要的 PBMC 细胞类型和状态,且其频率符合预期,共识别出代表主要 PBMC 群体的 13 个免疫聚类。为实现高分辨率的免疫表型分析,研究人员使用更大的探针面板(Xenium Prime 5K 人类泛组织和通路面板)对另外 75 万个细胞进行了STAMP分析,生成了循环中 31 种免疫细胞状态的高分辨率图谱,为跨时间(如年龄)或遗传(如种族背景)等维度的大规模图谱项目奠定了基础。值得注意的是,在分析中识别出 8 个 CD4+T 细胞亚群,从 naive 到效应和记忆群体,包括代表适应性免疫反应三个分支的 Th1、Th2 和 Th17 功能细胞类型;在 CD8+T 细胞库中也注释出 8 个亚群,从 naive 到中央 / 效应记忆群体以及不同类型的效应群体,包括干扰素反应性 CD8 T 细胞;在循环 NK 细胞库中,区分出 CD56dim CD16bright 和 CD56bright CD16dim NK 细胞,它们具有不同的抗体依赖性细胞毒性和迁移特性,同时还发现了一小部分增殖的 EOMES 和 CD34 表达细胞,可能代表 NK 祖细胞。尽管由于面板中缺少 Ig 相关基因和其他与 B 细胞表型分析相关的标志物,限制了对 B 细胞分化状态的深入注释,但在髓系群体中仍识别出明确的单核细胞亚群(经典型、中间型和非经典型单核细胞),并能够对树突状细胞(DCs)进行详细表征。
为评估 STAMP 技术的性能,研究人员将 STAMP-X(Xenium Prime 5K 人类泛组织和通路面板)与单细胞测序方法在转录本和基因检测灵敏度方面进行了基准测试。在分析两种方法共有的 4,808 个基因时,STAMP-X 显示出与常用的 3'、5' 和 Flex 单细胞实验(10× Genomics)相当的检测水平。由于基因面板设计的限制,与基于测序的实验的全转录组覆盖范围相比,STAMP 在分析所有基因时灵敏度较低。然而,按总基因数(靶向 / 捕获)归一化后,STAMP 的性能甚至超过了单细胞实验,表明其具有较高的分子检测效率。最后,在检测谱系定义的标志物基因(包括转录因子和表面标志物)方面,STAMP 与单细胞实验相当或更优。
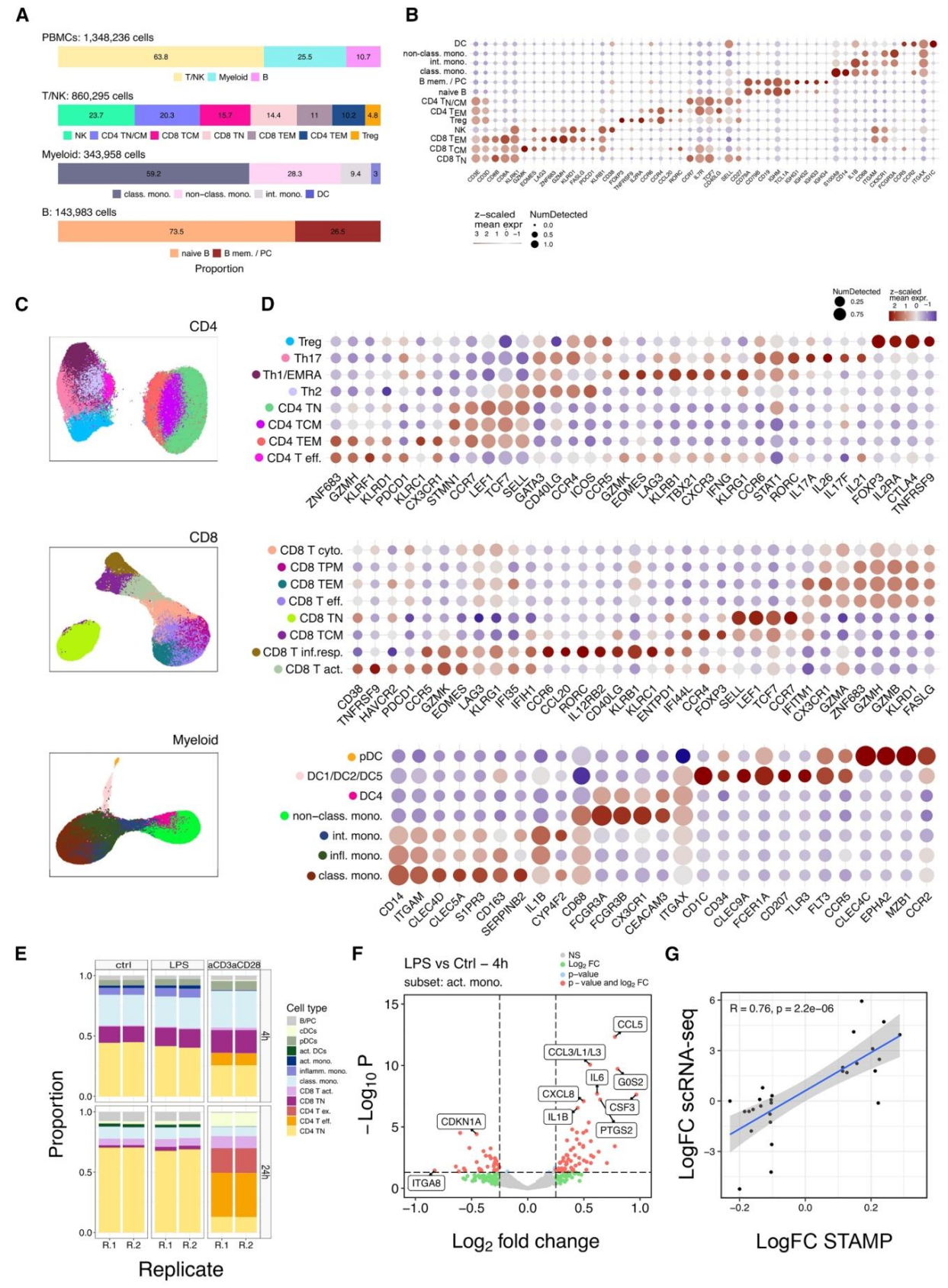
图4. 数百万循环血细胞的免疫表型分析
(五)免疫细胞扰动实验的多重分析
为评估 STAMP 技术在多重扰动研究中的适用性,研究人员将 PBMCs 分别在单独培养基(对照)、LPS(脂多糖)或 CD3/CD28 磁珠中培养,并在 4 小时和 24 小时收获细胞。使用 CosMx 1000-plex 人类通用细胞表征 RNA 面板进行分析,每个细胞的中位数转录本数为 320(范围:33-1,630),基因数为 183(范围:33-55),中位数细胞面积为 78μm²。过滤掉低质量细胞(18%)后,使用 InSituType(IST)无监督聚类识别出 12 个不同的免疫细胞聚类,与使用 Xenium 免疫肿瘤学面板获得的结果一致,捕获到 B 细胞、T 细胞和髓系细胞,其比例分别为 4%、68% 和 28%。
抗 CD3/CD28 刺激在 4 小时和 24 小时时间点逐渐减少 naive CD4+T 细胞,增加效应和耗竭 CD4+T 细胞;在 24 小时时, naive CD8+T 细胞也显著减少,而活化的 CD8+T 细胞数量增加。T 细胞活化还改变了髓系细胞的比例,经典单核细胞和浆细胞样 DCs(pDCs)在 4 小时时增加,而炎性单核细胞减少;在 24 小时时,常规 DCs 扩增,活化的 DCs 相对于对照减少。LPS 刺激引起的组成变化较为轻微,这与之前的报道一致。已知 LPS 通过细胞表面 Toll 样受体 4(TLR4)复合物激活单核细胞,相应地,在髓系细胞亚型中,活化的单核细胞显示出最高的 TLR4 表达水平。在 LPS 处理 4 小时后,活化单核细胞中有 39 个基因差异表达(30 个上调,9 个下调;FDR<0.05 且 log₂FC>0.25),其中包括促炎细胞因子和趋化因子基因(如 IL1B、IL6、CCL3、CCL5 和 CXCL8)以及其他与炎症相关的转录本(如 PTGS2、STAT4 和 CSF3)。到 24 小时时,活化单核细胞中 DUSP1 和 B2M 的上调以及 IL1B 的下调表明从早期炎症反应向限制炎症的调节反馈机制转变。
研究人员还使用 CellChat 探索了 LPS 激活后的细胞间通讯,重点关注 CXC 通路,因为它是炎症条件下诱导的主要分泌信号通路之一,通过白细胞迁移控制免疫反应。与单核细胞的 TLR4 介导的激活一致,检测到炎性和活化单核细胞与 CD4 效应 T 细胞之间存在强烈的信号传导,其中 CXCL8/CXCR1 受体-配体对是这些细胞类型之间通讯的主要贡献者,这些细胞类型也表达最高水平的趋化因子(CXCL8)及其同源受体(CXCR1)。最后,研究人员使用独立的 LPS 刺激 PBMCs 的单细胞测序数据集验证了 STAMP 技术识别的处理效应,发现两种实验中差异表达基因之间存在显著相关性(Pearson 相关系数 = 0.76,p<0.01)。
(六)干细胞分化过程中细胞状态动态的分析
为评估 STAMP 技术在识别 hESCs(人胚胎干细胞)响应 BMP4 处理分化过程中出现的细胞状态方面的分辨率,研究人员利用 STAMP 的多重分析能力,将八个 BMP4 处理时间点(0-120 小时)结合在一个 STAMP-C 实验中,使用 CosMx 人类通用细胞表征 RNA 面板。然后应用 Palantir 进行伪时间轨迹分析,以模拟 hESCs 响应 BMP4 处理的分化路径。
在该系统中,由 BMP 通路活性的存在与否决定的初始轨迹分支受到细胞在集落中位置的影响。由于 BMP 受体的基底外侧定位,大集落中心的细胞无法接触到培养基中的 BMP4。但由于该样本中大型集落相对较少,大多数经 BMP4 处理的 hESCs 在暴露于 BMP4 后 6 小时就从多能状态转变为中间 BMP 诱导状态,并持续到 12-24 小时,这种转变以 BMP4 通路靶基因(如 GATA3)的快速上调以及多能性标志物 SOX2 的下调为标志。
到 48 小时时,出现了中内胚层样状态(以 EOMES 和 KDR 表达为特征)和强烈表达 GATA3 的早期羊膜样状态。72 小时后,中内胚层样细胞分化为早期中胚层样细胞(表达 SNAI2 和 PDGFRA)、内胚层样细胞(表达 CXCR4 和 APOA1)和原始生殖细胞样细胞(PGCLC;表达 NANOG 和 CXCR4);早期羊膜样细胞分化为表达 TGFBI 的晚期羊膜样细胞,中胚层分支则进展为晚期中胚层样状态(以 DUSP6 和 FOXF1 上调为特征)。使用 Palantir,研究人员还计算了羊膜、中胚层和内胚层分支中关键标志物基因的表达动态,展示了多能性基因的逐步下调和谱系定义标志物的上调。对来自相同样本的细胞使用 Single-Cell Gene Expression Flex 实验进行处理,进行了相同的分析,结果显示出相似的轨迹,并且两种实验在不同分化时间点的基因表达具有强烈的正相关性。这些轨迹和标志物基因动态与之前基于 hESC 的原肠胚形成模型和原肠胚形成人类胚胎的 scRNA-seq 研究一致。
诱导多能干细胞(iPSCs)是通过细胞重编程从分化的供体细胞(如皮肤成纤维细胞)生成的多能细胞,可分化为各种细胞类型,是模拟人类发育和遗传疾病的宝贵工具,已广泛应用于疾病建模、药物筛选和毒性测试、再生医学、免疫研究以及谱系规范研究等领域。为探索 STAMP 技术在 iPSCs 研究中的潜力,研究人员将 iPSCs 分化为(神经)外胚层和中胚层谱系,并使用 Single-Cell Gene Expression Flex 实验和 STAMP-C(使用 CosMx 1000-plex 人类通用细胞表征 RNA 面板)同时进行 scRNA-seq 分析。降维和 PCA 显示,在两种实验中,前两个主成分都能区分未分化的亲本细胞、外胚层和中胚层。与亲本 iPSCs 相比,外胚层样本中神经外胚层标志物(SOX2 和 NRG1)的表达增加,多能性标志物(POU5F1 和 FGF2)的表达减少;中胚层细胞中中胚层相关标志物(PDGFRA、FOXF1 和 WNT5A)的表达上调。在 STAMP-C 中分析的每个细胞培养物的基因特征与在相同固定 iPSCs 悬浮液上生成的 scRNA-seq 数据集高度相关。
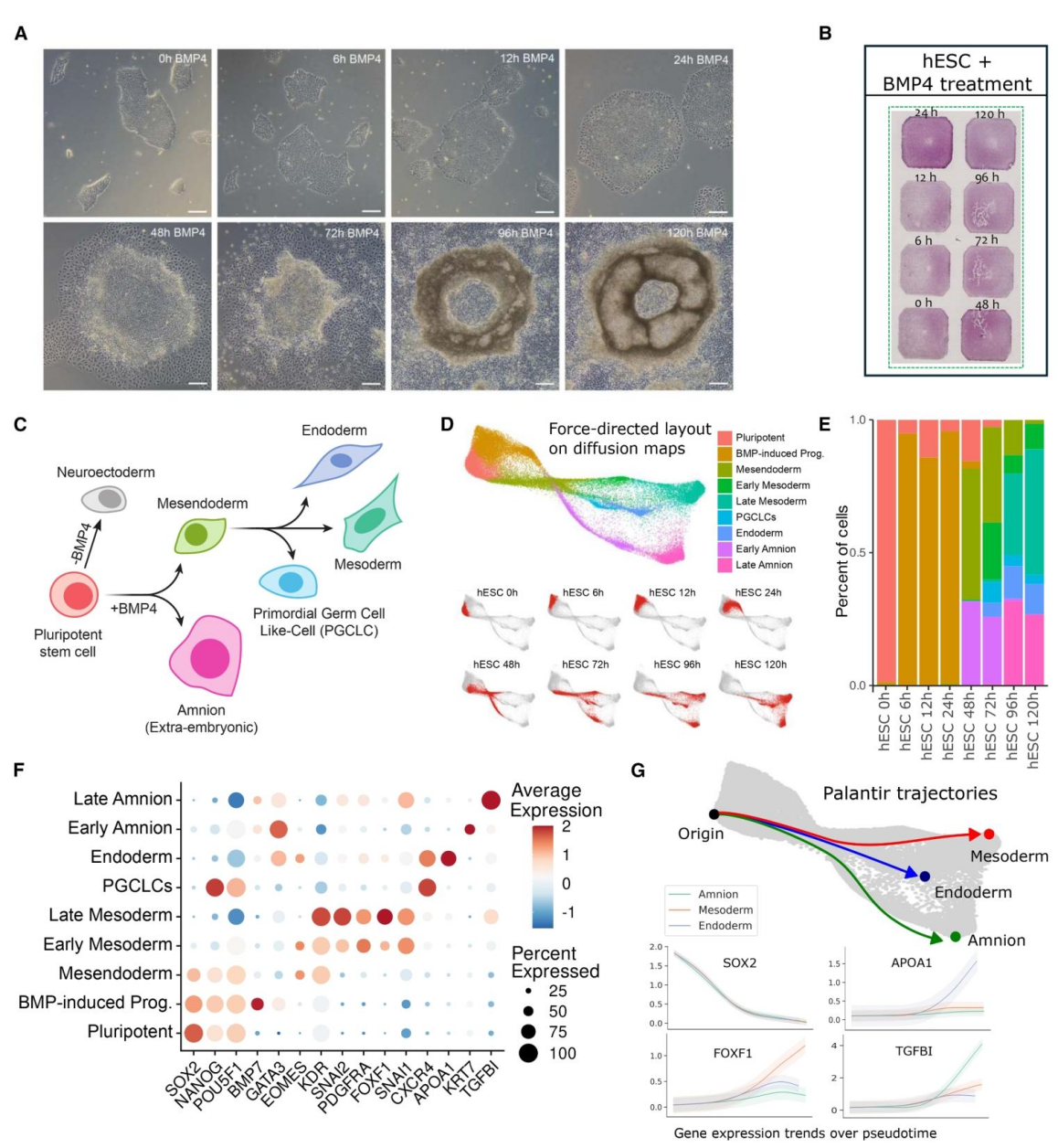
图 5. 干细胞分化过程中的细胞状态动态分析
(七)稀有细胞类型的灵敏检测
鉴于 STAMP 技术的可扩展设计,研究人员模拟了临床相关场景,特别是循环肿瘤细胞(CTCs)的识别。CTCs 对于量化肿瘤负荷、识别肿瘤异质性和检测可操作的改变具有重要价值。为模拟从血液样本中检测 CTCs 并评估 STAMP 技术在该实验设置中的灵敏度,研究人员将 MCF-7 癌细胞以 1:100,000 和 1:50,000 的稀释比例掺入 PBMC 样本中,在同一张载玻片上(总细胞数为 110 万个),然后使用 CosMx 人类通用细胞表征 RNA 面板对细胞混合物进行成像。
CosMx 细胞分割标志物提供了基准,可通过视觉检测 CTCs 模拟物,这些模拟物在每个 sub-STAMP 中随机分布(PCK 染色)。为实现 CTCs 模拟物的自动化检测,研究人员使用在固定细胞上进行的 scRNA-seq 生成基因表达特征,并根据 MCF-7 或 PBMC 谱对每个成像的单细胞进行评分。此外,结合 PCK 平均荧光强度、转录本计数数量和细胞面积的信息,能够以高准确度共同识别 CTCs 模拟物。在含有 10 个和 20 个目标掺入细胞的 sub-STAMP 中,研究人员分别可靠地识别出 7 个和 28 个 CTCs 模拟物,这些 CTCs 模拟物占总样本量的 0.001%,证明了 STAMP 技术检测具有临床应用潜力的极稀有细胞类型的能力。值得注意的是,免疫荧光图像允许研究人员根据与 STAMP 实验注册的坐标手动验证识别出的 CTCs 模拟物。这种稀有事件的检测与平台、面板或分割方法无关,通过使用 Xenium 免疫肿瘤学面板和 Xenium Prime 5K 人类泛组织和通路面板识别 CTCs 模拟物得到了证明,例如在含有 852,677 个 PBMCs 的样本中识别并确认了两个癌细胞的存在,展示了 STAMP 技术检测稀有细胞的灵敏度。
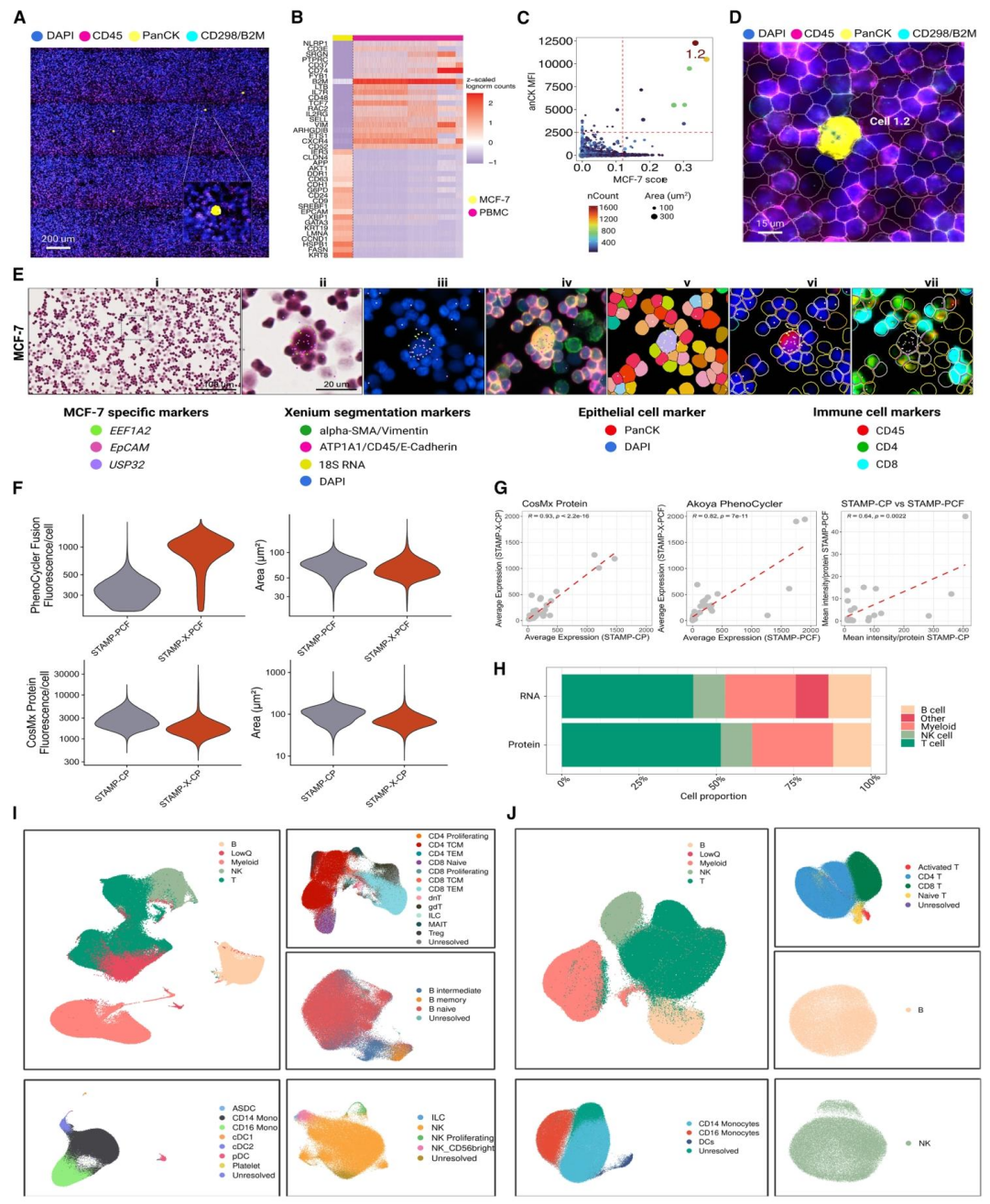
图 6. 循环肿瘤细胞模拟物与多模态分析
(八)RNA 与蛋白质结合的多模态分析
在基于成像的单细胞转录组学可扩展方法的基础上,研究人员进一步将 RNA 和蛋白质结合读数整合到 STAMP 框架中,以实现多模态细胞分析。为此,制备了两个高密度 STAMP(约 75 万至 90 万个 PBMCs),用于生成和整合 RNA 和蛋白质成像数据。首先进行 STAMP-X 分析,使用 Xenium Prime 5K 人类泛组织和通路面板进行高分辨率 RNA 分析,然后将同一张载玻片用于后续在两个不同平台上的蛋白质分析:CosMx SMI(64 个蛋白质面板;STAMP-X-CP)和 Akoya Phenocycler Fusion(40 个蛋白质面板,STAMP-X-PCF)。同时,在 CosMx SMI(STAMP-CP)和 Phenocycler Fusion(STAMP-PCF)上进行了平行的单模态蛋白质 STAMP 实验,以评估先前的 STAMP-X 转录组学处理对蛋白质分析结果的影响。
所有蛋白质实验都产生了高质量的蛋白质谱,每个细胞具有相当的平均荧光强度,证明了在 STAMP 框架内蛋白质检测的稳健性。对于 STAMP-CP,检测到 582,087 个细胞,平均荧光强度为 1,430.7,平均细胞面积为 77.25μm²;在 STAMP-X-CP 样本中,检测到 548,800 个细胞,平均荧光强度为 2,022,平均细胞面积为 73.62μm²。对于 Phenocycler,STAMP-PCF 样本包含 615,566 个细胞,平均强度为 350.8,平均细胞面积为 70.57μm²;STAMP-X-PCF 样本包含 630,691 个细胞,平均强度更高(923.1),平均细胞面积为 65.17μm²。
为进一步评估多模态分析的质量,研究人员比较了 STAMP-CP/X-CP 和 STAMP-PCF/X-PCF 样本(即有无先前 RNA 分析)中每种蛋白质的平均蛋白质表达。结果显示,两种平台上的蛋白质检测都显著相关(STAMP-CP/X-CP:R=0.93,p<0.01;STAMP-PCF/X-PCF:R=0.82,p<0.01)。值得注意的是,在单模态和多模态工作流程之间,特定蛋白质存在显著差异。为评估两种蛋白质组学技术之间的一致性,研究人员量化了共有蛋白质(n=20)的平均荧光强度,Pearson 相关分析显示出显著的正相关性(R=0.64,p=0.0022),其中 STAMP-CP 的总体平均荧光强度(最大值 = 407.35)高于 STAMP-PCF(最大值 = 46.96)。
研究人员还将来自 CosMx(-CP、X-CP)和 PhenoCycler Fusion(-PCF、-X-PCF)的 PBMCs 的 STAMP 蛋白质结果与基于测序的读数(即 CITE-seq)进行比较。CITE-seq 数据集包含 228 种抗体,其中 11 种与 CosMx 面板重叠,20 种与 PhenoCycler 面板重叠。正如预期的那样,由于面板尺寸更大,CITE-seq 每个细胞检测到的特征更多,当按面板尺寸归一化或分析特定谱系定义的标志物蛋白质时,这种影响有所减弱。为验证单模态和多模态工作流程中的 RNA 和蛋白质数据是否可整合用于跨模态免疫表型分析,研究人员对来自多模态 STAMP-X-CP 和 STAMP-X-PCF 的 PBMCs 进行了聚类分析,在两种模态中都识别出主要的细胞类型。对主要免疫细胞类型的转录组数据进行亚聚类,揭示了 26 个 T 细胞、B 细胞、自然杀伤(NK)细胞和髓系细胞亚群;蛋白质分析识别出了相同的主要细胞类型,但由于蛋白质面板较小,难以描述更细微的细胞状态。
(九)使用 STAMP 分析解离组织
尽管空间转录组学具有变革性潜力,但解离细胞的单细胞分析仍然是表征组织的金标准。为评估 STAMP 技术在解离组织中的通用性和适用性,研究人员对五种主要小鼠器官(肾脏、肝脏、大脑、心脏和肺)进行了组织分析,并选择这些器官是因为它们具有多样的细胞组成,可用于评估不同组织结构的分析效果。对于每个器官,都制备并处理了细胞和细胞核悬浮液,纳入细胞核是因为其制备可提供较少偏差的细胞类型覆盖范围,避免与细胞分离相关的转录 artifacts,并且与存档的冷冻标本兼容。将细胞和细胞核悬浮液固定在与 Xenium 兼容的玻璃载玻片上进行 STAMP-X 分析,并使用 Xenium Prime 5K 小鼠泛组织和通路面板进行分析。此外,还使用 Single-Cell Gene Expression Flex 实验对一部分固定细胞和细胞核进行了分析,以比较 STAMP 技术与已建立的 scRNA-seq 协议的质量和性能。总共在两个 STAMP-X 实验中分析了 601,417 个细胞(215,578 个细胞和 385,839 个细胞核)。
来自细胞和细胞核的 STAMP 数据在高质量细胞 / 细胞核的频率以及检测到的转录本和基因数量方面表现出可比性。在所有器官中,细胞和细胞核之间也观察到强烈的相关性(R=0.73,p<0.01),但肝脏在所有指标上都是一个明显的例外。正如先前报道的那样,从肝脏组织中分离单细胞面临重大挑战,这主要是由于该器官复杂的结构、肝细胞的脆弱性以及在肝脏酶高度活跃的温度下进行的当前解离方法的局限性。研究人员还纳入了大脑单细胞和单核制备物,因为它们在捕获高度连接的细胞类型(如神经元)方面存在明显偏差。在大脑细胞核上应用 STAMP 技术的效果优于单细胞制备,共获得 6,862 个细胞和 45,487 个细胞核,这与先前的发现一致。
使用小鼠参考细胞图谱进行高级注释,研究人员在所有器官中识别出 44 种不同的细胞类型,其中肺数据集包含最多的细胞类型(n=15),包括各种类型的 pneumocytes。在含有所有器官细胞 1:1:1:1:1 混合物的 sub-STAMP 中,研究人员通过应用源自每个相应组织的基因特征,在计算上分离了细胞身份。具体而言,对每个器官进行了一对一的差异表达分析,并验证了top差异表达基因的器官特异性。
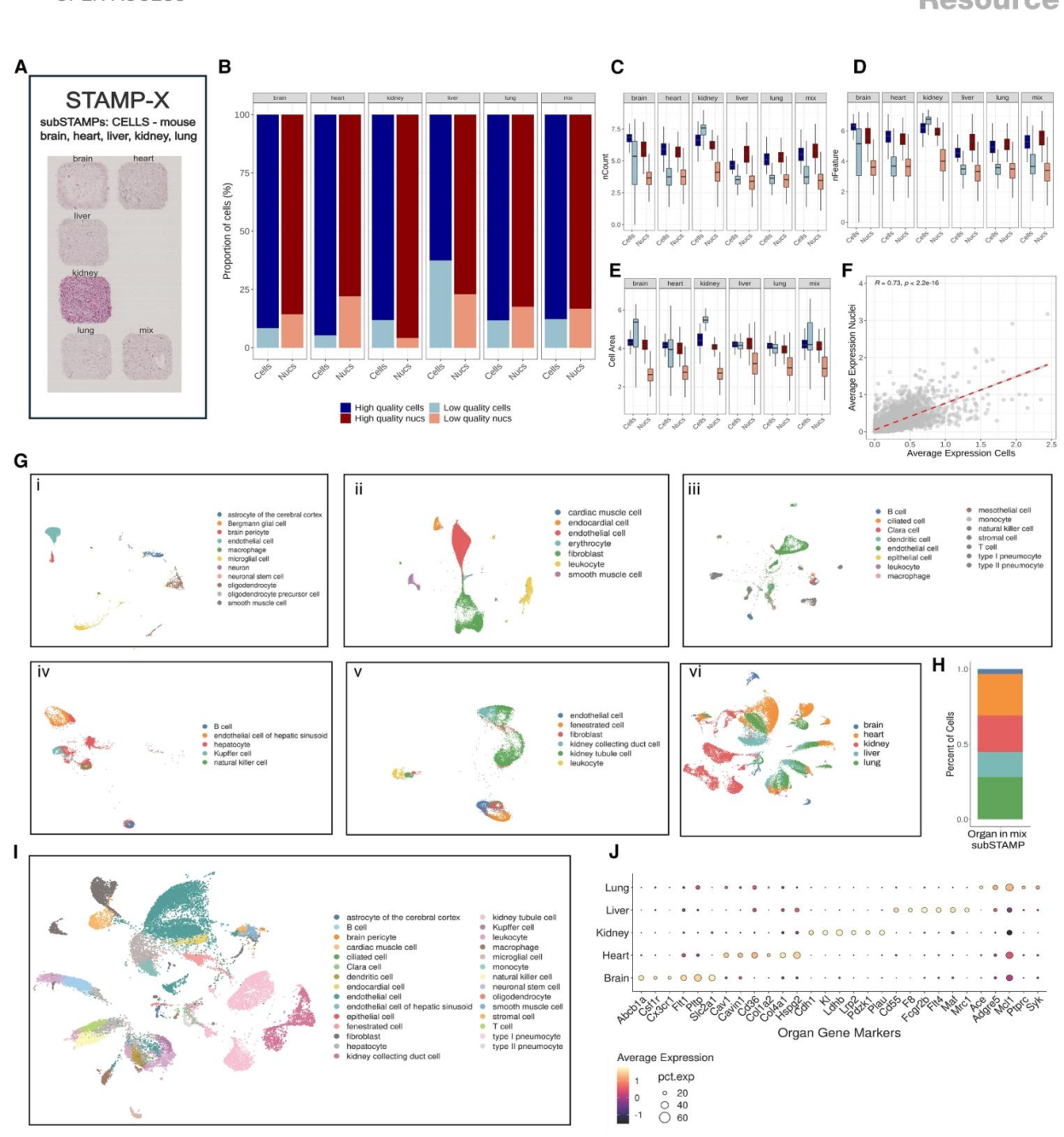
图 7. 利用 STAMP 技术分析解离组织
至此,我们总结下STAMP的特点:
(一)STAMP 技术的优势
总结下来STAMP主要有以下优势:
- 成本效益显著:相比单细胞测序,STAMP 每个细胞和每个特征的检测成本大幅降低,尤其适合大规模队列研究。
- 检测灵敏度高:在基因检测灵敏度上与单细胞测序相当,并可通过扩展探针集进一步提高分辨率。
- 多模态整合能力强:能够同时整合RNA、蛋白质和形态学信息,减少假阳性并支持多层次功能映射。
- 适用性广泛:可应用于多种样本类型(细胞系、PBMCs、干细胞、组织等)和研究场景(细胞类型识别、稀有细胞检测、免疫扰动实验等)。
(二)STAMP 技术的局限性
- 基因发现受限:依赖预定义基因面板,难以发现新基因和未知转录本。
- 存在杂交偏差:基于探针的检测可能导致表达谱失真,影响结果准确性。
- 不支持全长转录本分析:无法解析可变剪接事件,对转录本结构研究有限。
- 特定分子分析受限:在SNP、突变以及免疫受体(TCR/BCR)检测方面能力不足。
- CRISPR 筛选支持不足:仅能应用于小规模实验,不适合大规模基因功能筛选。
乐备实在单细胞与空间多组学研究开发中具有丰富的样本经验、分析经验。无论是探索复杂的生物学机制,还是推动新药研发,我们都有能力为客户提供全面、精准的科研服务。如果您也在寻找多技术联用的解决方案,欢迎各位老师同学与我们联系合作。








 沪公网安备31011502400759号
沪公网安备31011502400759号
 营业执照(三证合一)
营业执照(三证合一)




