







2025 年 8 月,《Circulation Research》在线发表明尼苏达大学 Xavier S. Revelo 团队的突破性研究。该研究证实,CCL24 可通过结合成纤维细胞表面受体 CCR3,经下游 PI3K/AKT 信号通路直接激活成纤维细胞;而成纤维细胞中 CCR3 的遗传敲除,能在压力过载模型中显著改善心脏功能并减轻纤维化程度。研究明确,CCL24/CCR3 信号轴的激活在压力超负荷诱导的心脏纤维化与心功能障碍进程中发挥关键调控作用。
研究背景:
炎症是心血管疾病发生发展的关键危险因素,通过调控机体对心脏损伤的适应性与非适应性应答,推动疾病进展。巨噬细胞作为心脏组织中丰度最高的免疫细胞,在心肌重塑过程中发挥核心调控作用;其中,心脏驻留巨噬细胞是心肌组织的重要组成部分,参与炎症反应启动、组织损伤修复及重塑的全过程。然而,心脏驻留巨噬细胞调控心力衰竭相关重塑的具体分子机制尚未完全阐明。
基于上述研究背景,本研究聚焦 CCL24 在急性压力超负荷诱导心力衰竭进展中的作用。研究发现,无论是 CCL24 基因缺陷模型,还是通过抗体介导的 CCL24 抑制策略,均能显著改善心脏功能,促进适应性心脏重塑,并减轻压力过载后的心肌纤维化程度。

研究结果:
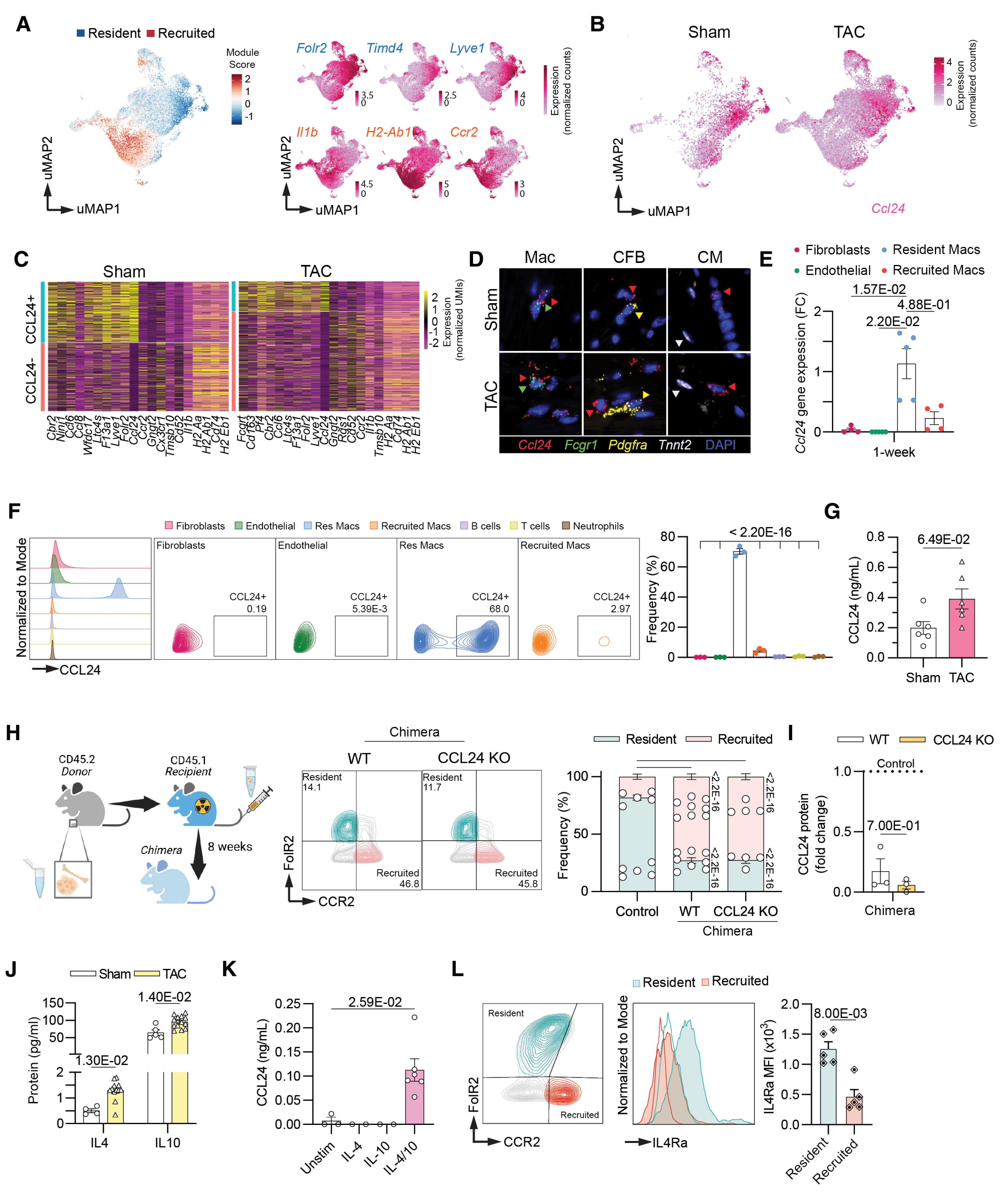
结论 1:心脏驻留巨噬细胞是心脏中 CCL24 的核心来源
本研究证实,心脏驻留巨噬细胞(CRMs)为心脏组织中 CCL24 的主要产生细胞。结合单细胞测序、qPCR、荧光原位杂交及骨髓移植实验结果验证,CCL24 的表达具有高度细胞特异性,主要富集于 CRMs,而非单核细胞来源巨噬细胞或其他心脏细胞亚群。在压力超负荷(TAC)模型中,心脏组织 CCL24 水平于心脏重塑早期呈短暂升高趋势,且与 CRMs 的扩增进程同步;进一步机制探究显示,IL-4 与 IL-10 可协同诱导 CRMs 分泌 CCL24,提示 CRMs 可能通过分泌 CCL24 在早期心脏重塑中发挥促纤维化作用。
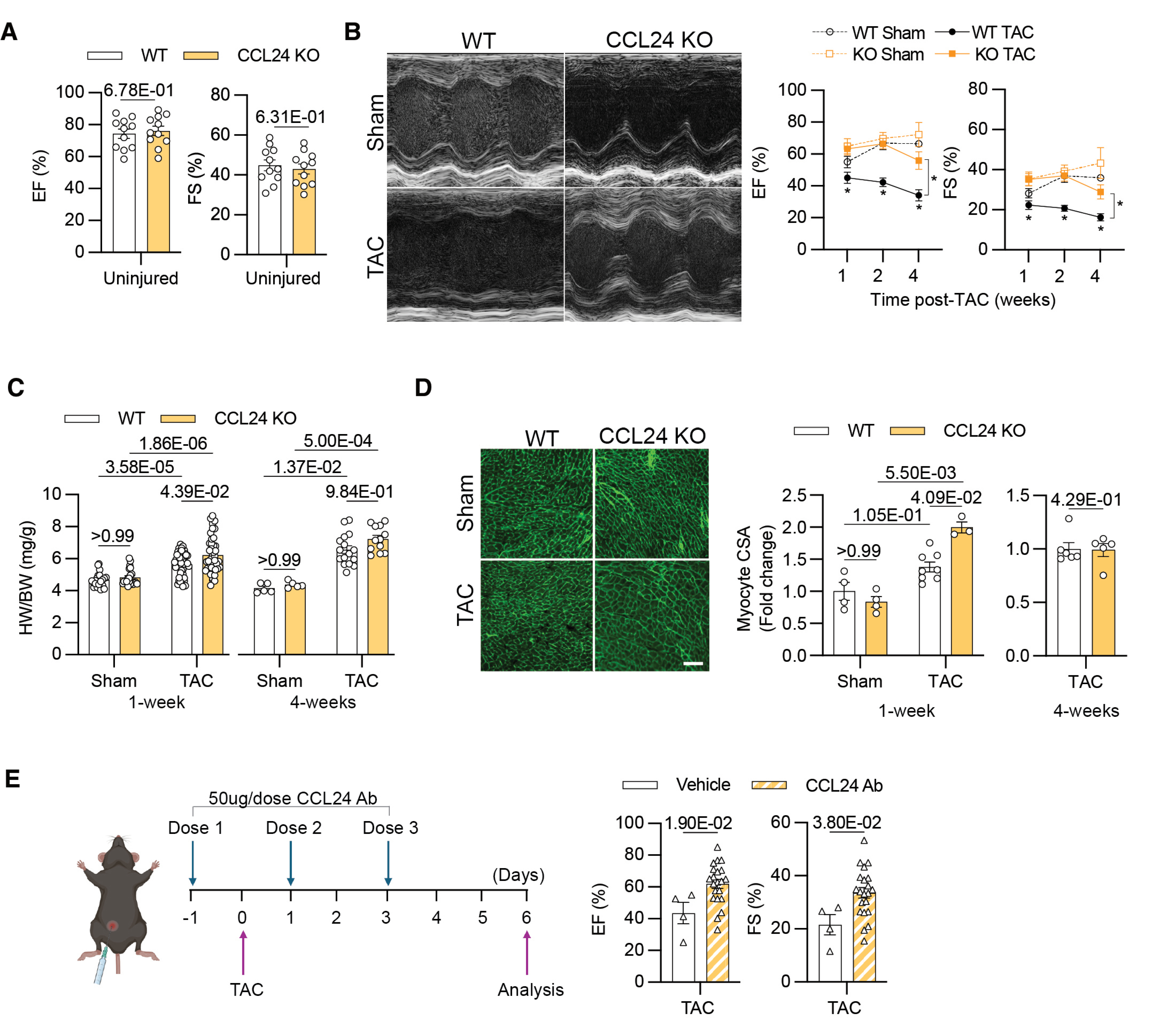
结论 2:心脏驻留巨噬细胞源性 CCL24 驱动心脏收缩功能障碍
压力超负荷条件下,CCL24 基因缺失或抗体中和干预均可显著改善小鼠心脏收缩功能,有效抑制心室扩张,且心肌肥大反应呈一过性特征。结合细胞溯源结果,介导该效应的 CCL24 来源于 CRMs 而非单核细胞来源巨噬细胞(MoMF),其持续存在会阻碍心脏适应性重构进程。以上结果提示,靶向阻断 CCL24 有望成为心力衰竭早期干预的潜在新策略。
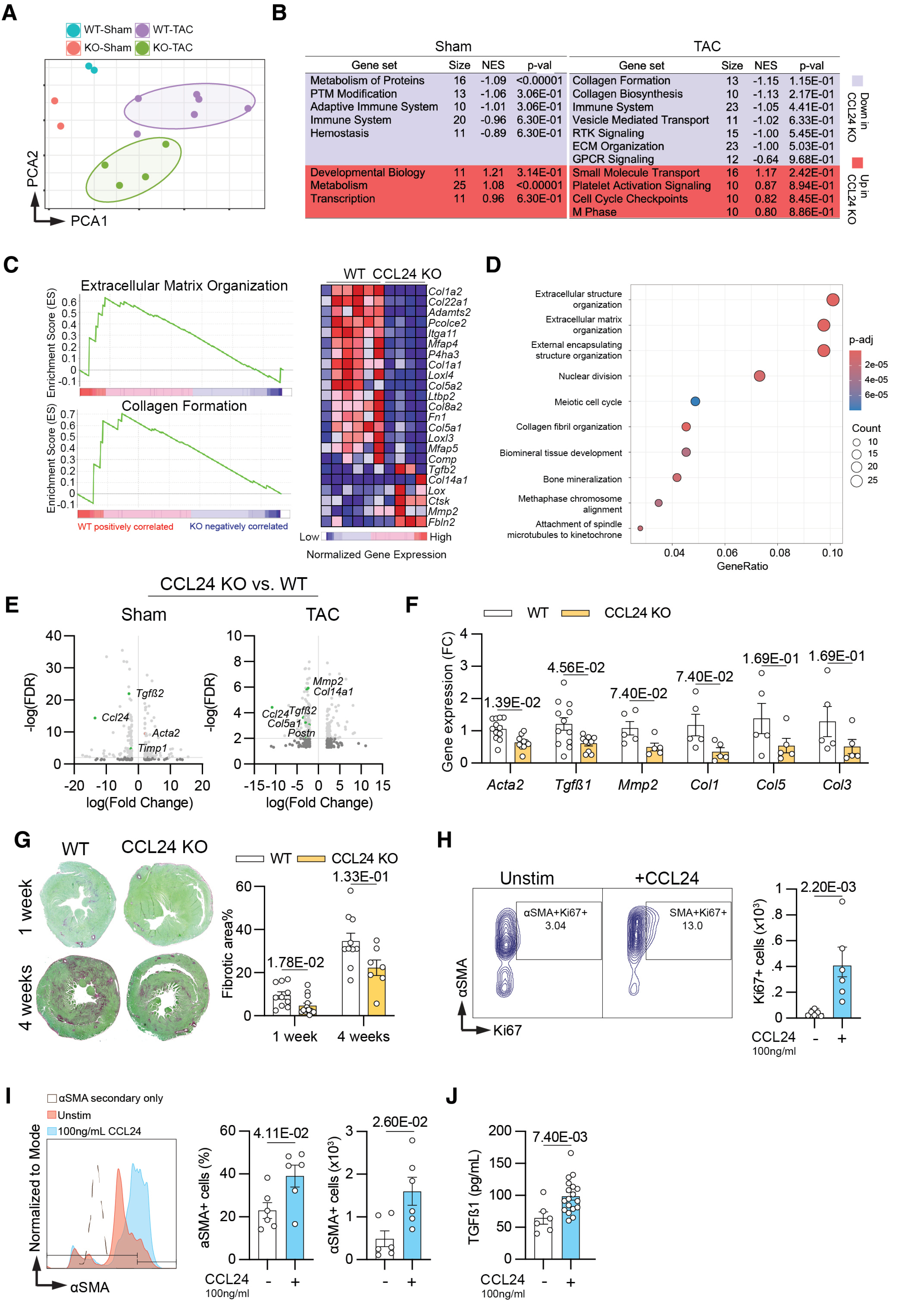
结论 3:CCL24 缺失可抑制心脏纤维化,且 CCL24 可直接激活成纤维细胞
RNA-seq 测序分析结果显示,CCL24 缺失可显著下调胶原合成、细胞外基质(ECM)形成相关基因表达,同时抑制 TGF-β 信号通路激活,使促纤维化基因表达谱整体下调,最终导致心脏纤维化面积减少 40%。体外功能验证实验证实,重组 CCL24 蛋白可直接诱导成纤维细胞增殖,并促进 α- 平滑肌肌动蛋白(αSMA)及 TGF-β 的产生,明确 CCL24 通过直接激活成纤维细胞驱动心脏纤维化进程。
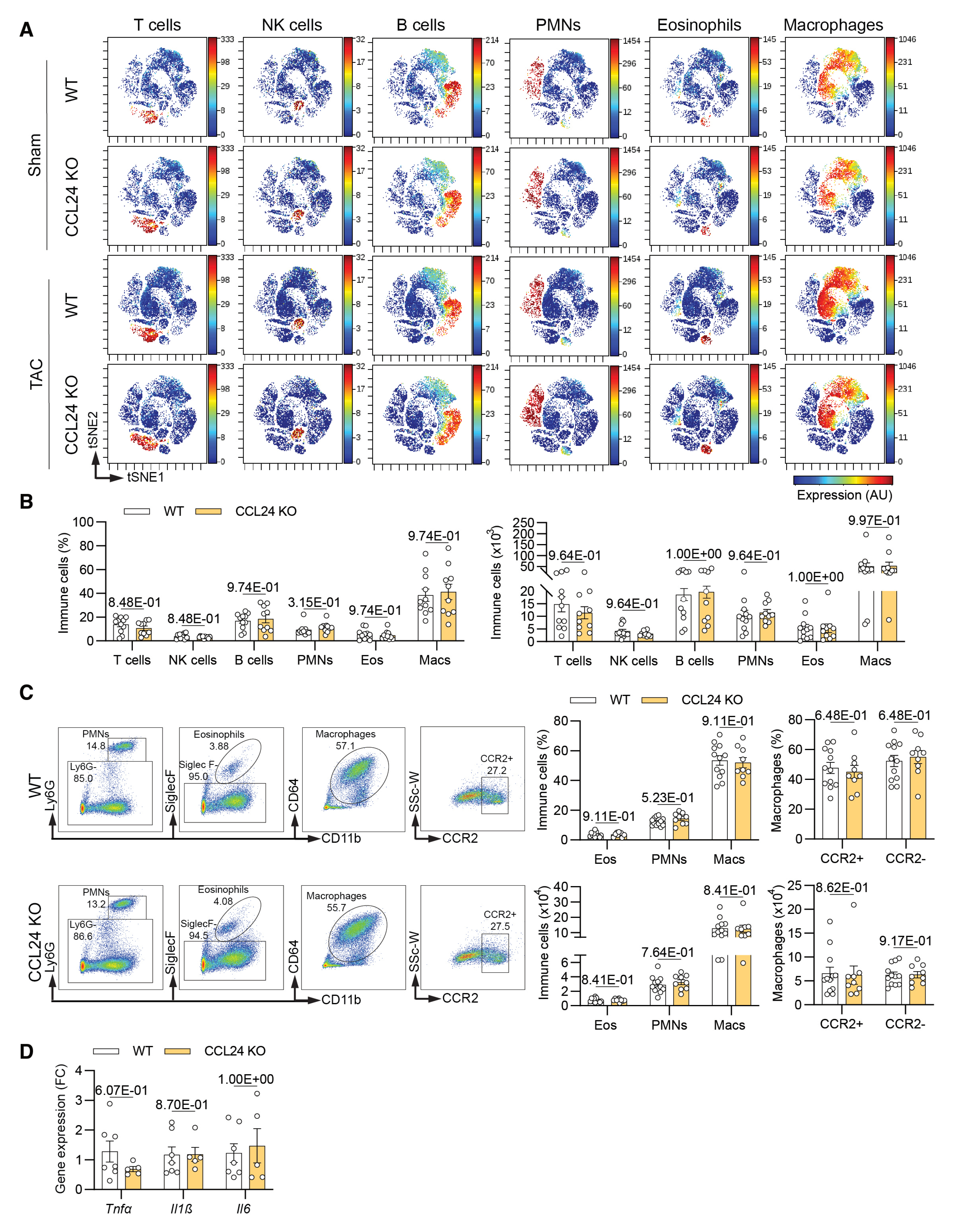
结论 4:CCL24 介导的纤维化重塑独立于心脏炎症反应
进一步研究发现,CCL24 缺失对稳态状态及压力负荷后的心脏组织中各免疫细胞亚群数量无显著影响,炎症相关基因表达水平也未发生明显改变。该结果提示,CCL24 调控心脏纤维化重塑及心功能障碍的作用机制,与心脏局部炎症细胞浸润及炎症反应强度变化无关,具有炎症非依赖性特征。
结论 5:CCL24 通过 CCR3 受体介导促纤维化效应
机制探究证实,CCL24 可通过特异性结合心脏成纤维细胞表面的 CCR3 受体,直接激活成纤维细胞;并通过诱导 TGF-β 依赖的信号通路,促进胶原及 αSMA 的表达,最终驱动心肌纤维化。功能阻断实验显示,无论是靶向抑制 CCR3 受体活性,还是阻断 TGF-β 信号,均可有效抑制上述纤维化进程,显著改善心脏功能及间质纤维化程度。该结果明确 CCL24-CCR3 信号轴是调控心脏纤维化的关键通路,具备靶向干预的临床潜力。
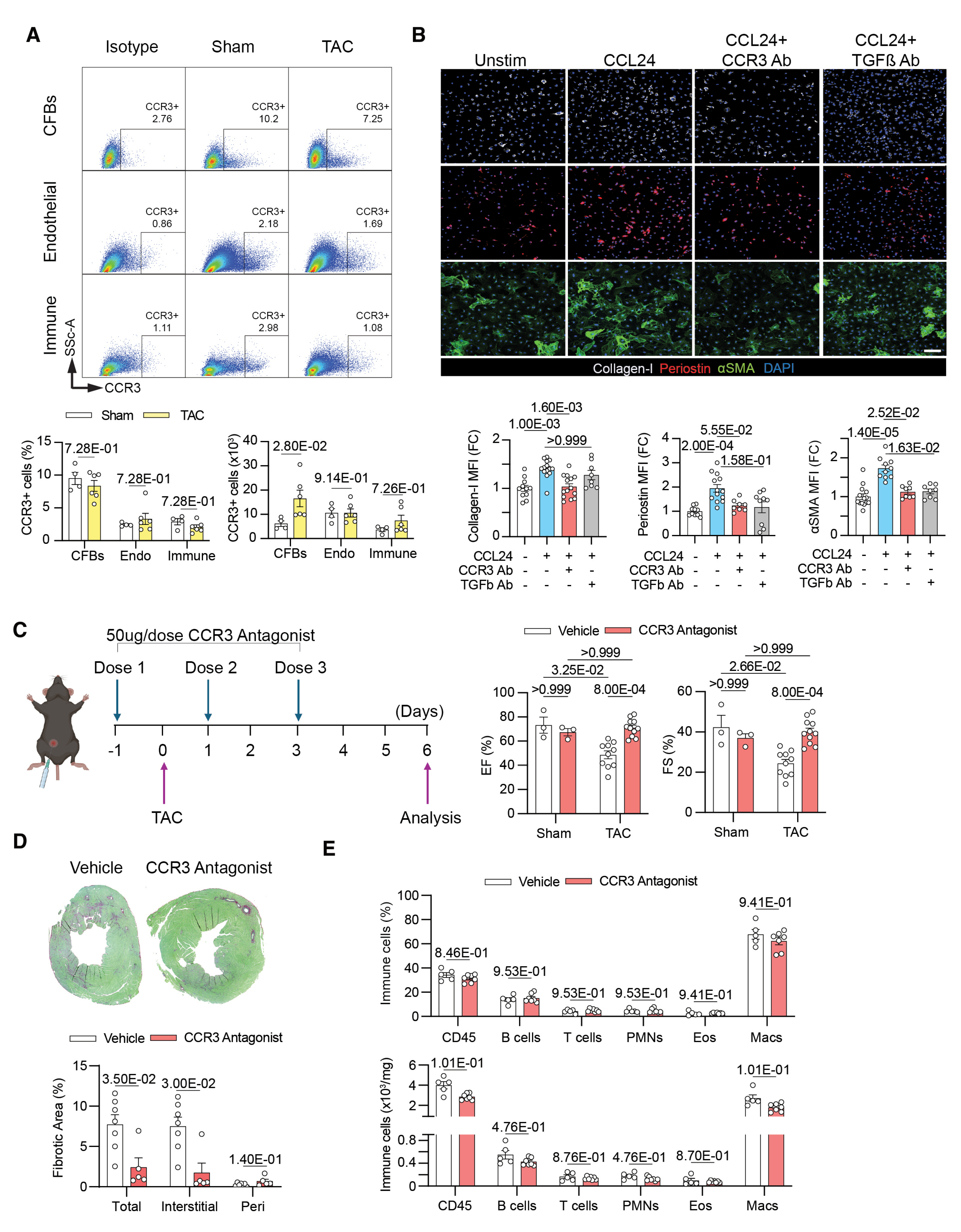
结论 6:成纤维细胞特异性 CCR3 条件性敲除可抑制心脏纤维化进展
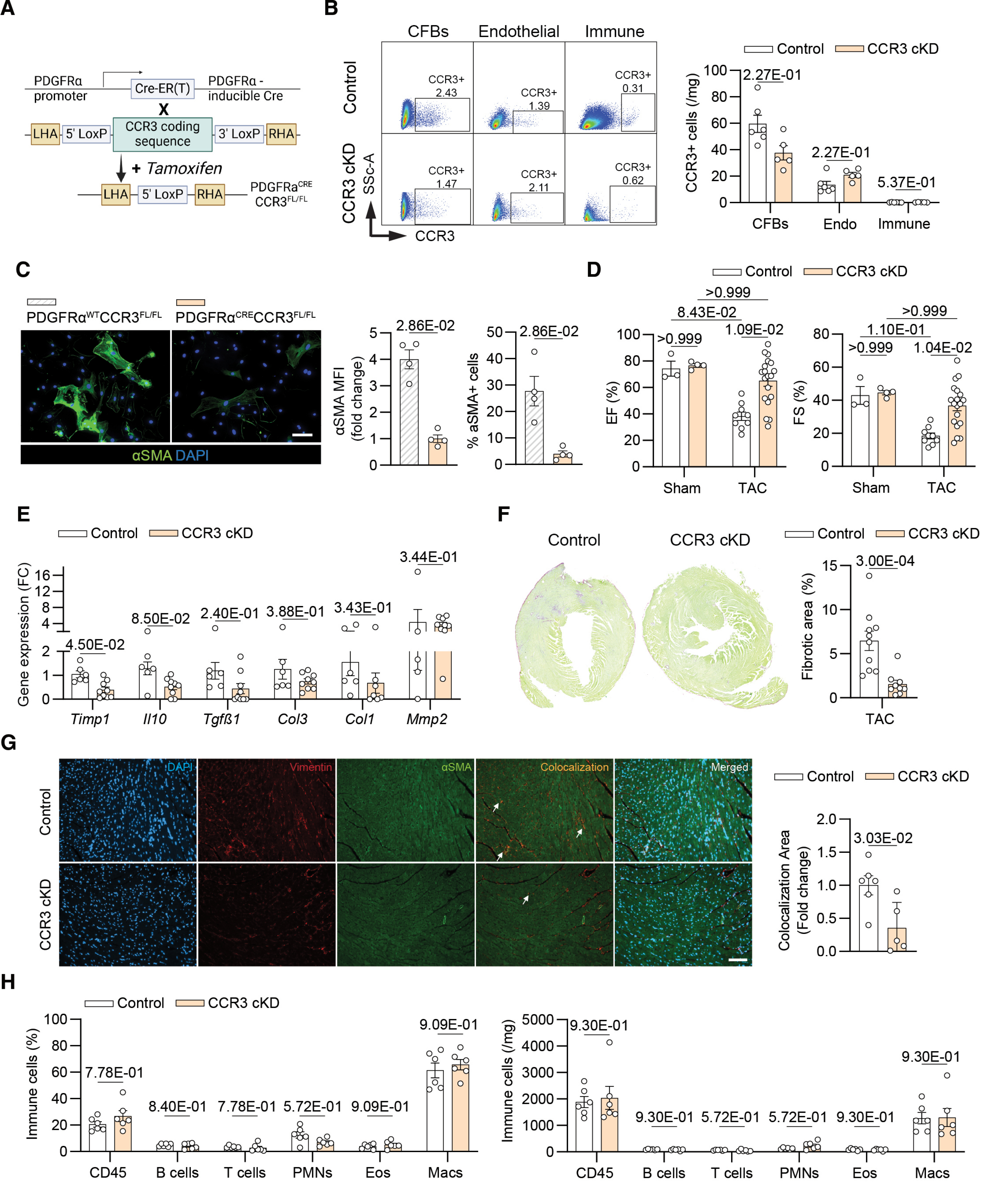
本研究构建成纤维细胞特异性 CCR3 条件性敲除小鼠(CCR3 cKD),经他莫昔芬诱导后,小鼠成纤维细胞中 CCR3 表达水平显著下调。在主动脉缩窄术(TAC)构建的压力超负荷模型中,术后 1 周检测显示,CCR3 cKD 小鼠的左心室射血分数(LVEF)和左心室缩短分数(LVFS)较对照组显著提升;纤维化相关标志基因(Col1、Col3、Timp1)表达显著下调,心肌组织胶原沉积量及肌成纤维细胞(α-SMA 阳性细胞)面积明显减少,心功能得到有效改善。进一步分析证实,上述心功能改善与纤维化抑制效应不依赖心脏组织免疫细胞浸润状态的改变,明确成纤维细胞表面的 CCR3 是介导 CCL24 促心脏纤维化作用的关键分子靶点。
结论 7:CCL24-CCR3 轴通过激活 PI3K/Akt 信号通路驱动心脏纤维化
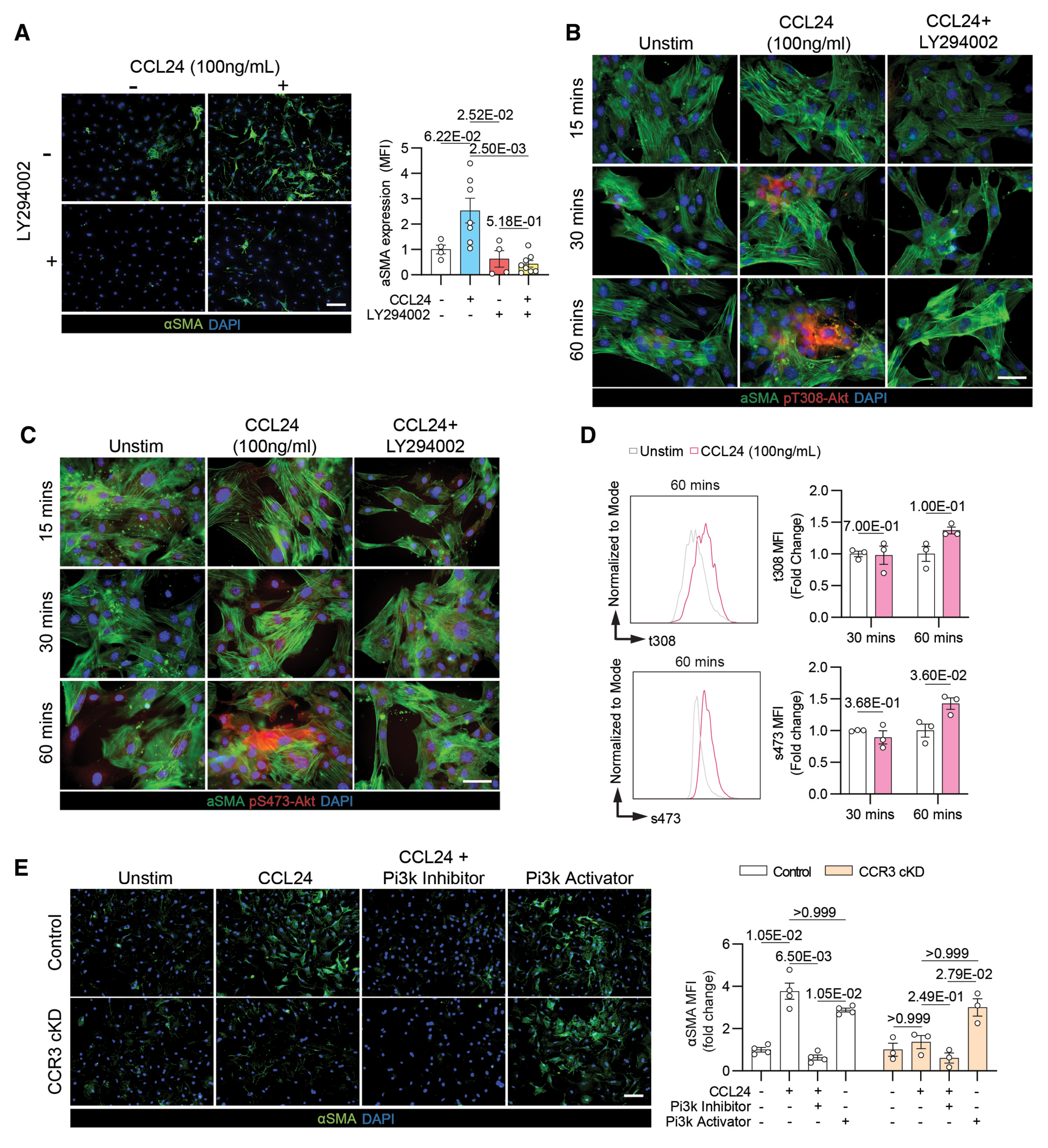
机制探究明确,CCL24 与成纤维细胞表面 CCR3 结合后,可特异性触发下游 PI3K/Akt 信号通路激活:体外实验显示,CCL24 刺激 30-60 分钟可诱导 Akt 蛋白磷酸化水平显著升高;而使用 PI3K 特异性抑制剂或敲除 CCR3,均能有效阻断 CCL24 诱导的 α-SMA(肌成纤维细胞活化标志)表达上调。功能挽救实验进一步证实,直接激活 PI3K 信号可绕过 CCL24 的调控,直接诱导肌成纤维细胞活化。上述结果共同证实,PI3K/Akt 信号通路是 CCL24-CCR3 轴介导心脏纤维化的核心下游调控通路。
总结
本研究明确,在压力超负荷病理状态下,心脏驻留巨噬细胞通过分泌 CCL24,特异性结合成纤维细胞表面的 CCR3 受体,激活下游 PI3K/Akt 信号通路,进而促进 TGF-β 释放,驱动心肌纤维化进程,最终加重心功能障碍。功能干预实验证实,无论是基因层面的 CCL24-CCR3 轴缺陷,还是药物介导的靶向阻断,均能显著抑制心肌纤维化并改善心脏功能。基于上述结果,本研究提出靶向 CCL24-CCR3 轴可作为心力衰竭早期抗纤维化的潜在新策略,但该策略的长期干预效果及临床应用安全性仍需进一步验证。
参考文章:
Parthiban, Preethy et al. “Macrophage-Derived CCL24 Promotes Cardiac Fibrosis Via Fibroblast CCR3.” Circulation research vol. 137,9 (2025): 1140-1156. doi:10.1161/CIRCRESAHA.
CCL24趋化因子检测服务哪里有?
乐备实提供CCL24等多因子检测服务:
| 货号 | 产品名 | 检测指标 |
| LXLBH40-1 | 人趋化因子/细胞因子-40因子Panel | 6Ckine/CCL21,BCA-1/CXCL13,CTACK/CCL27,ENA-78/CXCL5,Eotaxin/CCL11,Eotaxin-2/CCL24,Eotaxin-3/CCL26,Fractalkine/CX3CL1,GCP-2/CXCL6,GM-CSF,GRO-α (Gro-a/KC/CXCL1),Gro-β/CXCL2,I-309/CCL1,IFN-γ,IL-1β,IL-2,IL-4,IL-6,IL-8/CXCL8,IL-10,IL-16,IP-10/CXCL10,I-TAC/CXCL11,MCP-1/CCL2,MCP-2/CCL8,MCP-3/CCL7,MCP-4/CCL13,MDC/CCL22,MIF,MIG/CXCL9,MIP-1α/CCL3,MIP-1δ/CCL15,MIP-3α/CCL20,MIP-3β/CCL19,MPIF-1/CCL23,SCYB16/CXCL16,SDF-1α+β/CXCL12,TARC/CCL17,TECK/CCL25,TNF-α |
| LXLBM31-1 | 小鼠趋化因子-31因子Panel | BCA-1/CXCL13,CTACK/CCL27,ENA-78/CXCL5,Eotaxin/CCL11,Eotaxin-2/CCL24,Fractalkine/CX3CL1,GM-CSF,I-309/CCL1,IFN-γ,IL-1β,IL-2,IL-4,IL-6,IL-10,IL-16,IP-10/CXCL10,I-TAC/CXCL11,KC/CXCL1,MCP-1/CCL2,MCP-3/CCL7,MCP-5/CCL12,MDC/CCL22,MIP-1α/CCL3,MIP-1β/CCL4,MIP-3α/CCL20,MIP-3β/CCL19,RANTES/CCL5,SCYB16/CXCL16,SDF-1α/CXCL12,TARC/CCL17,TNF-α |








 沪公网安备31011502400759号
沪公网安备31011502400759号
 营业执照(三证合一)
营业执照(三证合一)




