







一、PI3K的组成与激活机制
磷脂酰肌醇3-激酶(Phosphatidylinositide 3-kinases,PI3K)是一种胞内磷脂酰肌醇激酶,同时具有丝氨酸/苏氨酸激酶活性。根据结构特征,PI3K可分为三类,其中研究最为深入的为I类PI3K。该类PI3K以异源二聚体形式存在,由一个调节亚基和一个催化亚基共同组成。
催化亚基包括α、β、δ、γ四种类型。其中,α、β、δ三类催化亚基主要与p85α、p85β或p55等调节亚基结合;而γ类催化亚基则与p101或p84/87调节亚基相结合。调节亚基含有SH2结构域,能够识别受体酪氨酸激酶(RTKs)的胞内激酶结构域,进而诱导催化亚基的活化。
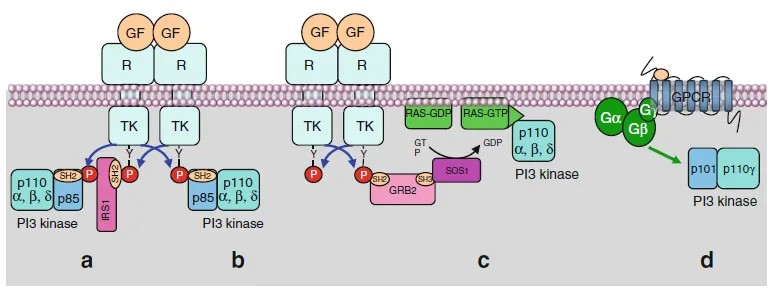
图 1. PI3K的激活方式
PI3K的激活主要涉及四种机制。如图1所示:第一,当胰岛素家族生长因子激活相应RTKs时,调节亚基可通过接头蛋白(如IRS1)间接被活化(a);第二,其他家族生长因子与RTKs结合后,调节亚基也可直接识别RTKs的酪氨酸激酶结构域而激活(b);第三,活化的RAS蛋白(RAS-GTP形式)能够直接激活PI3K的催化亚基(c)。RAS同时调节Raf和PI3K,构成MAPK通路与PI3K通路之间交叉对话的主要分子基础;第四,γ型PI3K催化亚基还可被部分G蛋白偶联受体直接激活(d)。
二、PI3K的催化功能
PI3K被激活后,可催化磷脂酰肌醇-4,5-二磷酸(PIP2)的3位羟基磷酸化,生成磷脂酰肌醇-3,4,5-三磷酸(PIP3),进而启动下游信号转导。相反,细胞内另一种酶——PTEN,则催化该反应的逆过程,将PIP3去磷酸化恢复为PIP2,从而负向调控PI3K介导的信号通路。
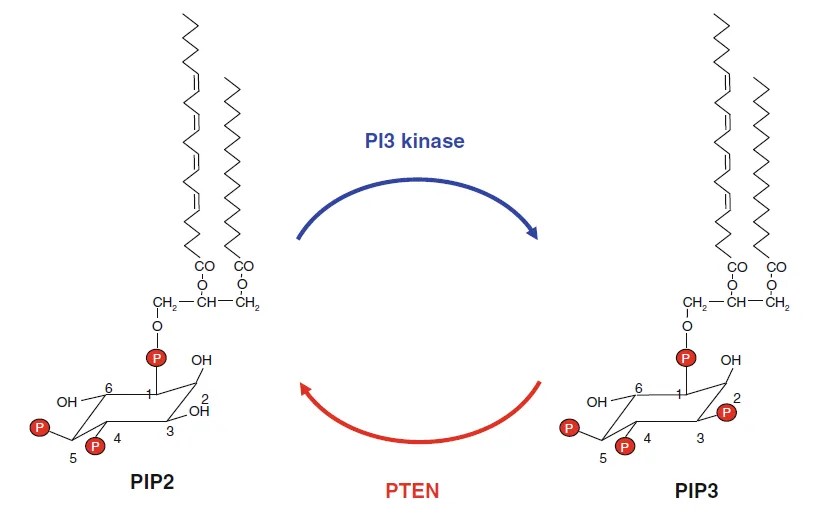
图 2. PI3K催化的反应
PIP3生成后作为第二信使,同时将PDK1与AKT蛋白招募至质膜。在质膜上,PDK1磷酸化AKT蛋白第308位苏氨酸残基(T308),促使AKT发生部分活化。活化的AKT进一步作用于下游多条调控通路,介导细胞生长、代谢及存活等生物学过程。
三、PI3K-AKT信号通路及其下游效应
AKT属于AGC家族丝氨酸/苏氨酸激酶,主要包含三个结构域:PH结构域(对PIP3具有亲和力,对膜结合至关重要)、催化结构域及调节结构域。AKT作为一种原癌基因,在葡萄糖代谢、细胞凋亡、细胞增殖、转录调控及细胞迁移等多种细胞过程中发挥关键作用。PI3K/AKT信号通路的失调见于多种人类疾病,包括癌症、糖尿病、心血管疾病及神经退行性疾病。
AKT下游作用的底物种类繁多。结合图3,对主要底物概述如下:
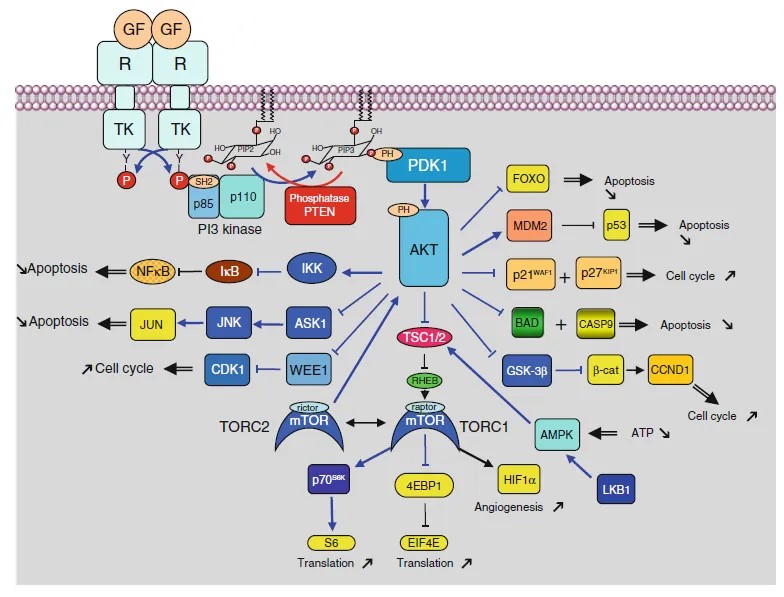
图 3. PI3K-AKT及下游蛋白
TSC2
TSC复合体包含TSC1、TSC2及TBC1D7三个亚基,可负向调控mTORC1复合体活性,发挥肿瘤抑制作用。其机制在于:TSC2含有的GTP酶活化蛋白结构域可将GTP形式的RHEB水解为GDP形式,使RHEB失活,从而抑制mTORC1激酶活性,阻碍细胞生长。AKT磷酸化TSC2蛋白的Ser939及Thr1462位点后,TSC2活性受到抑制,进而导致mTOR通路被激活。
BAD
BAD可正向调节线粒体凋亡。AKT磷酸化BAD后,其活性受到抑制。
MDM2
AKT通过磷酸化p53的泛素连接酶MDM2而激活后者,从而抑制p53介导的细胞周期阻滞与凋亡。
p21与p27
CDK抑制因子p21与p27是重要的细胞周期调节分子。在正常细胞DNA受损时,它们被诱导产生,抑制DNA复制并增强DNA修复,发挥抑癌作用。AKT通过磷酸化抑制这些CDK抑制因子的活性。
GSK3β
GSK3β是一种进化上保守的丝氨酸/苏氨酸激酶,参与肝糖代谢及Wnt信号通路。在正常情况下,GSK3β可磷酸化β-catenin,使其经蛋白酶体降解。AKT磷酸化GSK3β后,导致GSK3β失活,无法磷酸化β-catenin,使β-catenin在胞浆内大量聚集并进入细胞核,激活与细胞分裂及生长调控相关的基因(如c-myc与Cyclin D1)。
IKK
IKK被AKT磷酸化而激活。活化的IKK进一步磷酸化IκB(一种抑制NF-κB通路的转录因子),最终结果为正调控NF-κB通路,从而影响细胞生存、增殖、侵袭、血管生成及化疗耐药性。
WEE1
CDK1抑制激酶WEE1可被AKT磷酸化而抑制,从而影响有丝分裂进程。
ASK1
ASK1是一种可激活JNK信号通路的MAPKKK,与凋亡诱导、促凋亡及内质网应激相关。AKT磷酸化ASK1后,其活性受到抑制。
FOXO
FOXO是一种诱导凋亡的转录因子。AKT磷酸化FOXO后,导致FOXO在细胞内重新定位并丧失转录活性。
综上所述,AKT所产生的生物学效应总体上倾向于促进细胞增殖并抑制凋亡。
四、PI3K-AKT-mTOR通路及TORC复合物
如前所述,AKT可通过抑制TSC2间接促使RHEB激活mTOR。mTOR全称为哺乳动物雷帕霉素靶点,是一类丝氨酸/苏氨酸激酶。其C端与磷脂酰肌醇激酶催化结构域同源,但不具备磷脂酰肌醇激酶活性,而是具有丝氨酸/苏氨酸蛋白激酶活性。mTOR可形成两种复合物:TORC1与TORC2。
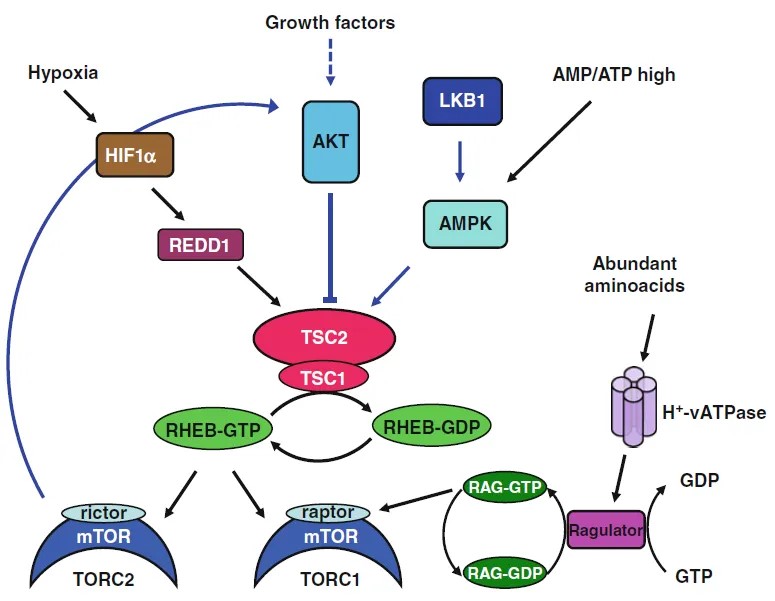
图 4. PI3K-AKT-mTOR通路
除受AKT激活外,mTOR还可根据细胞内的能量与营养状况被调控。如图4所示,当能量不足(AMP/ATP比例升高)或缺氧时,TSC1与TSC2形成复合体的能力增强,从而抑制mTOR复合物的形成。当溶酶体内氨基酸过量时,H⁺-ATP酶可激活含有鸟嘌呤核苷酸交换因子的Ragulator蛋白复合物,进而促进TORC1的形成。

图 5. TORC1与TORC2
如前所述,mTOR存在两种复合物:mTORC1与mTORC2。mTORC1由mTOR、Raptor、MLST8以及非核心组分PRAS40和DEPTOR组成。mTORC2由mTOR、RICTOR、MLST8和mSIN1组成。两者主要区别在于:mTORC1含有Raptor,对雷帕霉素敏感;mTORC2含有RICTOR,对雷帕霉素不敏感。此外,PRAS40是AKT的底物,而mSIN1是MAPK通路的底物。
从功能上看,TORC2的作用相对专一,可磷酸化AKT,对mTOR形成正反馈调节,促进mTOR进一步活化。TORC1则功能更为广泛,可继续磷酸化下游多种底物,调控细胞生存、增殖、蛋白质合成、脂质生成、血管生成及自噬等生物学过程。
五、TORC1下游底物及其生物学效应
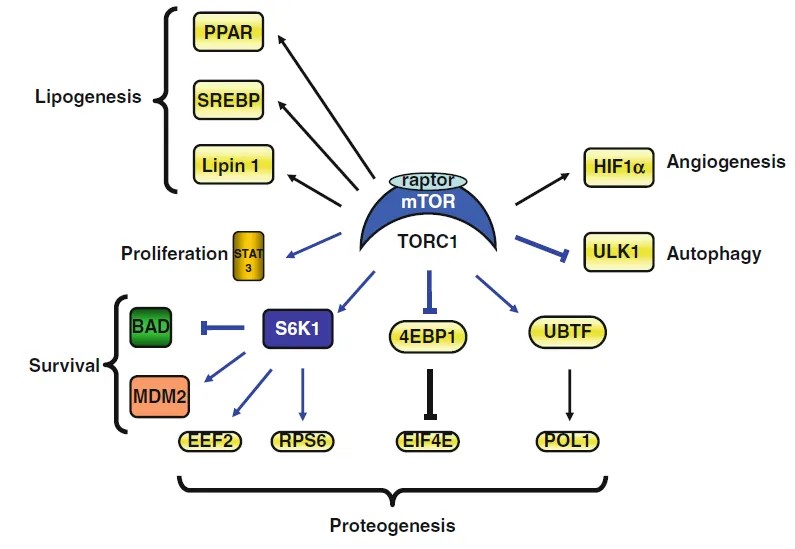
图 6. TORC1下游底物及效应
TORC1与蛋白质合成
真核起始因子4E是一种帽结合蛋白,能够特异性识别mRNA 5′端的帽子结构,在真核翻译起始过程中发挥关键作用。当其阻遏蛋白4EBP1处于低磷酸化状态时,eIF4E无法被释放,从而不能招募核糖体以启动翻译。TORC1通过磷酸化4EBP1,解除其对eIF4E的抑制作用,进而促进翻译及蛋白质合成。
TORC1还可先磷酸化激活下游的AGC家族核糖体S6激酶(S6K1)。S6K1可进一步磷酸化多种底物,包括核糖体S6蛋白(RPS6)、PDK1(AKT上游激活蛋白)、MDM2(p53抑制因子)、转录调节因子EEF2以及接头蛋白IRS等;同时,S6K1还可通过磷酸化抑制促凋亡蛋白BAD。上述效应共同促进蛋白质合成并抑制细胞凋亡。
TORC1与脂质合成
除促进蛋白质合成外,TORC1还可通过磷酸化激活STAT3促进细胞增殖;同时激活SREBPs以及PPARα和PPARγ,从而促进脂质合成。
TORC1与血管生成
TORC1能够通过激活HIF1α促进血管生成。值得注意的是,HIF1α也可间接激活TSC2,从而对TORC1产生负反馈调控作用。
TORC1与自噬
TORC1通过磷酸化抑制下游底物ULK1等自噬相关蛋白,从而负向调控自噬过程。
综上所述,TORC1通过磷酸化激活或抑制一系列下游底物,直接或间接促进蛋白质与脂质合成,推动细胞增殖,最终引起细胞与组织的生长。
六、PI3K-AKT-mTOR通路与肿瘤发生
如前所述,PI3K信号通路可通过多种机制抑制细胞凋亡、促进细胞存活与增殖。在肿瘤发生过程中,PI3K/AKT信号通路广泛处于激活状态,其中编码关键组分的基因如PIK3CA、PIK3R1、PTEN及AKT等存在高频突变。以PIK3CA基因为例,其在约36%的乳腺癌病例中发生突变,与肿瘤的发生、进展及耐药性密切相关。
目前该通路涉及的主要突变类型包括:
1. PIK3CA突变
PIK3CA基因编码PI3K催化亚基p110,在多种肿瘤中具有较高的突变频率。在前列腺癌、乳腺癌等癌症类型中,超过10%的病例存在PIK3CA突变,其中激活型突变占比超过80%。这些激活点突变可使PI3K蛋白不依赖上游信号而持续处于活化状态。PIK3CA突变被认为是EGFR-TKI治疗产生耐药性的潜在原因之一。
2. PTEN突变
如前所述,抑癌基因PTEN可拮抗PI3K的生物学效应,因此其突变亦促进肿瘤发展。在子宫内膜癌、脑癌、皮肤癌及前列腺癌中,超过10%的病例存在PTEN基因突变。
3. AKT突变
AKT基因家族包括AKT1、AKT2和AKT3。在肿瘤中,AKT最常见的突变形式为激活点突变及基因拷贝数异常扩增。在乳腺癌、结直肠癌、卵巢癌及膀胱癌中,AKT突变的比例均较高。








 沪公网安备31011502400759号
沪公网安备31011502400759号
 营业执照(三证合一)
营业执照(三证合一)




