







Cell Ranger 堪称 10X genomics 单细胞测序的 “黄金搭档” !作为官方配套分析软件,它能直接 “吃进” Illumina 原始数据(BCL 格式),一口气 “吐出” 表达矩阵、PCA 降维、聚类分析(Graph-based + K-Means 双模式)、t-SNE 可视化 全套结果,再搭配 Loupe Cell Browser ,直接开了 “单细胞数据探索盲盒”,想挖啥细节都方便!
因为 Cell Ranger 把复杂分析全 “打包” 了,单细胞测序数据探索门槛直接砍半 ,研究者再也不用卡在生信分析里熬秃头等结果,能把时间留给更有意思的生物学意义挖掘。
今天浅浅唠唠这些数据里藏着啥门道!
【Summary/ 概要】

- 有效细胞数(Estimated Number of Cells):实验捕获的活细胞总量,代表数据量基本盘。
- Mean Reads per Cell:总测序读数(Number of Reads)÷ 有效细胞数,反映单个细胞平均测序深度。
- Median Genes per Cell:所有细胞中,检测到基因数的中位数,体现细胞基因表达检测的覆盖度。
【Sequencing/ 测序】

-
Number of Reads:样本测序的reads总数。 - 测序数据量换算(以 PE150 测序为例):
测序数据量(G) = (reads总数 × 150 × 2) ÷ 10⁹。比如 reads 总数 3.39 亿,数据量≈101.76G 。 - 测序饱和度(Sequencing Saturation):被测到 ≥2 次的 reads 占比。核心逻辑:测序深度(目标区域重复测序次数)和文库复杂度(细胞质量、异质性、RNA 组成等)共同决定。文库越复杂(转录本种类 / 数量多),饱和越难;细胞数多但单个细胞 reads 少,那么饱和度低,反之则高。关键结论:饱和度越高,继续加测意义越小;曲线末端平滑≈接近饱和,加测难提基因检出数。
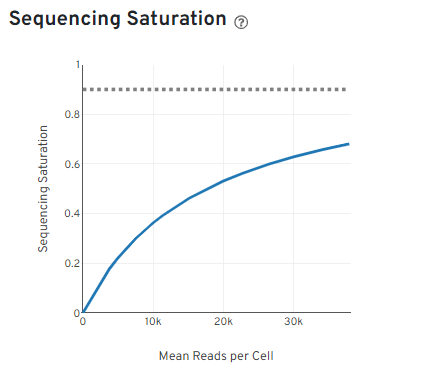
- 测序饱和度曲线终点附近的斜率表征增加测序深度时基因检测收益的理论上限,该斜率通过局部回归模型(LOESS)拟合计算。通常以虚线标记接近饱和点的临界值,当曲线末端趋于平缓(斜率绝对值<0.05),提示当前测序深度已接近文库复杂度上限,继续增加测序数据量对基因检测数的提升幅度将≤5%,此时可判定为测序饱和状态。该特征可通过 Shannon-Wiener 指数或 Good's coverage 指数进行量化验证,是优化测序成本与数据完整性的关键参考指标。
- Q30:测序质量核心指标!公式
Q=-10log₁₀(P)(P 是碱基错误率),Q30 意味着 “1000 个碱基里最多 1 个错”,数值越高质量越好。- Q30 Bases in Barcode/RNA Read/UMI:分别统计 barcode、RNA 读数、UMI 里质量分>30 的碱基占比,反映不同区域测序准确性。
- Barcode Q30 碱基比率:统计 barcode 区域中质量分数>30 的碱基占比,Q30 对应碱基错误率≤0.1%,该指标反映微流控建库中细胞标签的识别准确性。
- RNA Read Q30 碱基比率:针对 RNA 测序读段的碱基质量统计,Q30 比率越高表明转录本序列的可靠性越强,是过滤低质量测序数据的关键参数。
- UMI Q30 碱基比率:评估 UMI(唯一分子标识符)区域的碱基质量,高质量 UMI 是准确量化基因表达拷贝数的基础,可有效降低 PCR 扩增偏差的影响。
- 有效 Barcode reads 总数:经质量过滤后,包含完整且正确 barcode 序列的测序读段总数,直接关联单细胞分群的可靠性。
- 有效 UMI reads 总数:携带有效 UMI 序列的测序读段总数,用于基因表达量的绝对定量分析,其数值稳定性影响差异表达分析的可信度。
【Mapping】
-
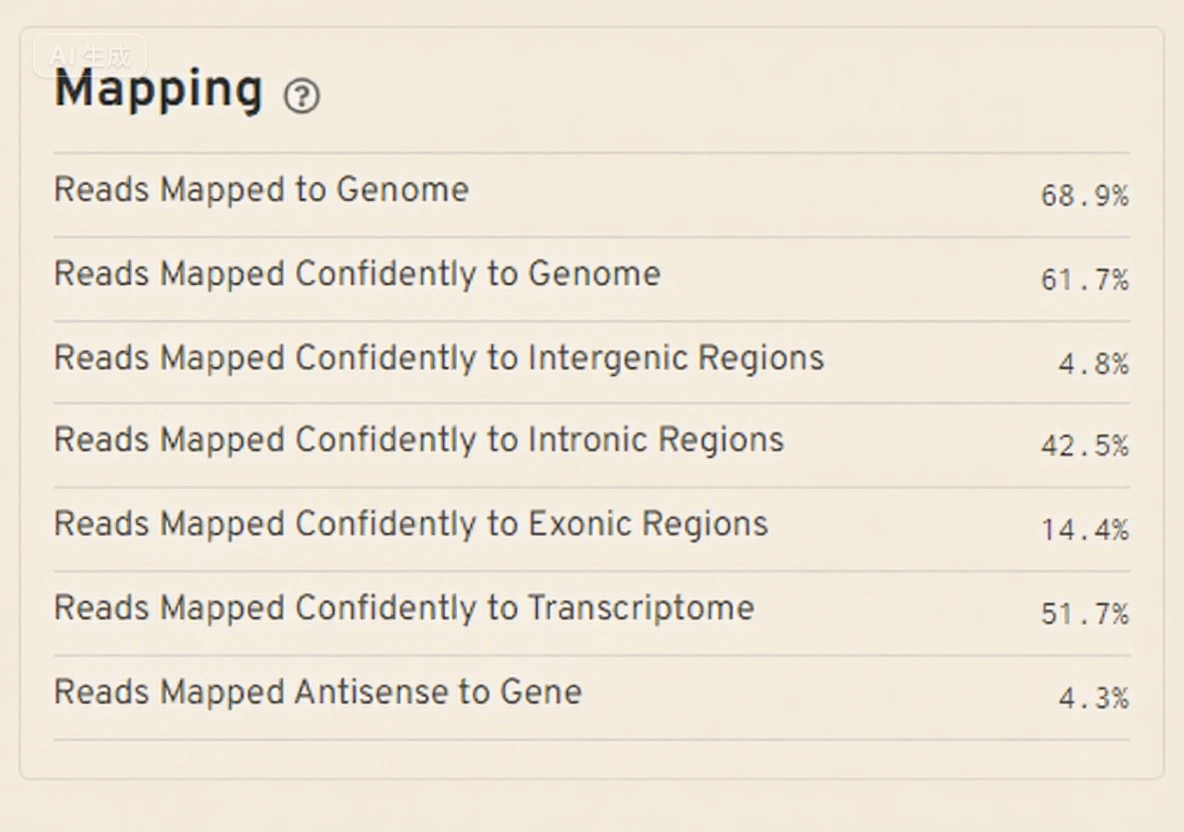
Reads Mapped to Genomes:比对到参考基因组上的 Reads 在总 Reads 中占的比例,人鼠样本一般要求≥80%。该指标反映测序数据与目标物种基因组的匹配程度。
-
Reads Mapped Confidently to Genome:比对到参考基因组并得到转录本 GTF 信息支持的 Reads 占比。若某条 Reads 同时可比对到外显子区与非外显子区,优先判定为外显子区比对(基于基因结构注释优先级)。
-
Reads Mapped Confidently to Intergenic Regions:比对到基因间区域(唯一基因间区)的 Reads 占比,反映非编码区序列的测序信号分布。
-
Reads Mapped Confidently to Intronic Regions:比对到内含子区域的 Reads 占比。细胞核实验中该比例通常高于细胞样本(核转录本丰富),是 RNA Velocity 分析的关键数据。
-
Reads Mapped Confidently to Exonic Regions:比对到外显子区域的 Reads 占比,直接关联 mRNA 成熟转录本的定量分析。
-
Reads Mapped Confidently to Transcriptome:比对到已知参考转录本的 Reads 占比,主要用于 UMI 计数统计,人鼠样本建议≥50%。
-
Reads Mapped Antisense to Gene:比对到基因反义链上的 Reads 比例,可能反映非编码 RNA 调控或转录本剪切异常等生物学现象。
【Cell/ 细胞】
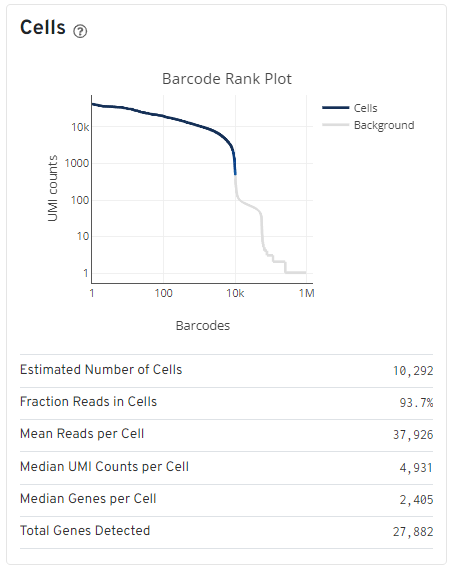

深蓝色曲线代表检测到的真实细胞数量分布,反映样本中具有完整转录组活性的细胞群体信号强度;灰色曲线为背景噪音信号,主要包含细胞碎片、游离 mRNA 及空包结构的测序信号。绿色箭头标记的蓝灰曲线分界点是细胞质量阈值,该阈值越靠上提示高质量细胞占比越高,反之则表明低质量组分(如细胞碎片、游离 RNA)比例增加。曲线交界处的显著骤降(斜率>10)是高质量细胞与背景区分度良好的核心特征。蓝色箭头标记的节点用来区分完整细胞和背景,在微流控 “油包水” 建库体系中,背景信号主要来源于两类结构:仅含胶珠与 barcode 引物的空包 GEMs,以及细胞死亡后释放、被空载 GEMs 捕获的游离 RNA。生物信息学通过 barcode reads 强度阈值区分细胞与背景 —— 真实细胞 GEMs 的 barcode reads 数通常为背景信号的 10 倍以上(基于泊松分布模型校正,10X Genomics Cell Ranger 软件默认设定为 10× 阈值)。Estimated Number of Cells 指基于 barcode 聚类算法估算的细胞总数,人鼠样本理想值需≥2000 个,用于评估样本捕获效率与细胞存活率。Fraction Reads in Cells 表示细胞相关 reads 占总测序 reads 的百分比,理想值>70%,该指标表征测序数据用于有效细胞分析的比例。当该数值<50% 时,需排查样本制备环节(如细胞解离导致的 RNA 释放)或建库体系(如 GEMs 包裹效率)。
背景 RNA 污染与细胞 RNA 含量的动态平衡直接影响细胞识别准确性。若背景 RNA 浓度升高(如样本冻融损伤)或细胞 RNA 含量降低(如低活性细胞群),会导致 barcode reads 阈值判别误差,建议结合单细胞转录本分布密度(Transcriptome UMI density)进行二次验证。
一、单细胞测序深度指标:
Mean Reads per Cell= 总测序读数(Number of Reads)÷ 有效细胞数(Estimated Number of Cells),反映单个细胞平均分配到的测序数据量,就像 “每个细胞的测序资源配额”,数值越高说明单个细胞的数据覆盖越充分。
二、基因表达丰度指标:Median Genes per Cell
- 定义:所有细胞中检测到基因数的中位数(即把细胞按基因数排序,中间值代表 “典型细胞” 的基因表达量)。
- 标准参考:
- 理想值≥700:基因数足够支撑细胞分群(比如免疫细胞、肿瘤细胞等不同类型能靠基因差异分开);
- <500 需警惕:数据可能因细胞质量差(如凋亡)或测序深度不足导致不可靠。
- 细胞类型影响:
- 低表达类:成熟 B/T 细胞(免疫细胞分化成熟后基因表达更 “专一”);
- 高表达类:肿瘤细胞、干细胞(基因活性强,表达谱更广泛)。
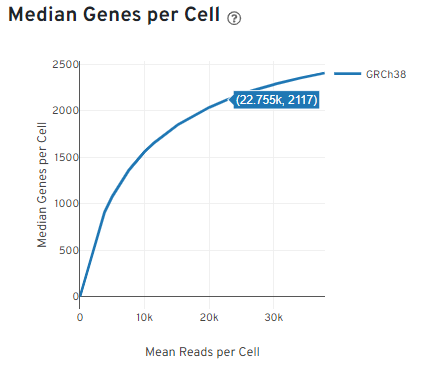
三、测序饱和度判断:基因中位数曲线
- 曲线意义:横轴是测序深度,纵轴是中位数基因数,终点斜率代表 “加测能多检出多少基因”。
- 关键信号:曲线末端变平滑→测序饱和,此时加测不会显著提升基因检测数,可停止测序节省成本~
四、全局基因覆盖指标:Total Genes Detected
= 至少被 1 条 UMI 标记的基因总数,直接体现测序对样本基因表达的 “扫描范围”,数值越大说明检测到的基因种类越全面(但需结合细胞数和测序深度综合判断)。
【Gene Expression/ 基因表达】
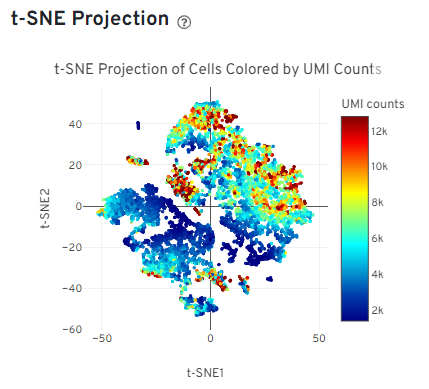
这是单细胞 UMI 定量与基因表达关联图谱:
- 横轴与纵轴:通过 t-SNE 算法将高维基因表达数据 “压缩” 成二维坐标,距离越近的细胞,基因表达模式越相似(就像把说同一种 “语言” 的细胞归到一起)。
- 点的特征:每个点代表一个细胞,点的大小或颜色对应细胞 UMI 总数(UMI 是标记单细胞 RNA 的分子标签)。UMI 数量越多,说明这个细胞的基因表达活性可能越强(就像 “话痨” 细胞会发出更多信号)。
核心应用:通过 UMI 分布和细胞聚类位置,能快速找到高表达细胞群、识别异常细胞(比如离群点),还能辅助判断细胞类型(同类细胞通常扎堆出现)。
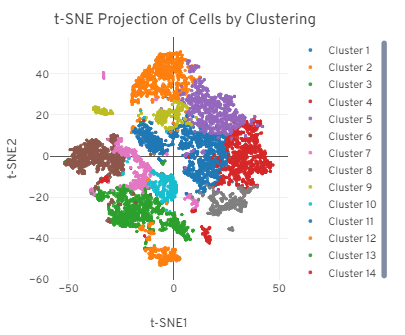
这是单细胞自动聚类图谱:
- 聚类原理:算法像 "基因表达侦探",把基因表达谱相似的细胞归到同一群(比如免疫细胞群、肿瘤细胞群),就像给细胞按 "语言口音" 分组。
- t-SNE 二维图:高维基因数据被 "压缩" 成横纵轴坐标,距离越近的细胞,表达模式越相似(就像邻居聊天更投机)。
- 显示规则:图中只随机展示部分细胞(全量显示会密密麻麻看不清),但聚类逻辑适用于所有细胞哦~
关键点:看不同颜色簇的分布 —— 紧凑的簇代表同类细胞,分散的点可能是稀有细胞或异常细胞,簇间距离越远说明细胞类型差异越大!
乐备实是国内专注于提供高质量蛋白检测以及组学分析服务的实验服务专家,自2018年成立以来,乐备实不断寻求突破,公司的服务技术平台已扩展到单细胞测序、空间多组学、流式检测、超敏电化学发光、Luminex多因子检测、抗体芯片、PCR Array、ELISA、Elispot、多色免疫组化等30多个,建立起了一套涵盖基因、蛋白、细胞以及组织水平实验的完整检测体系。
我们可提供从样本运输、储存管理、样本制备、样本检测到检测数据分析的全流程服务。凭借严格的实验室管理流程、标准化实验室操作、原始数据储存体系以及实验项目管理系统,已经为超过3000家客户单位提供服务,年检测样本超过100万,受到了广大客户的信任与支持。







 沪公网安备31011502400759号
沪公网安备31011502400759号
 营业执照(三证合一)
营业执照(三证合一)




