













一、研究背景与数据基础
随着高通量测序技术的发展,生物大数据为肿瘤分子机制研究提供了丰富资源。宫颈癌作为女性常见恶性肿瘤,其发病机制复杂,寻找可靠的预后标志物对临床诊疗具有重要意义。本文以 2019 年发表于《Journal of Cellular Physiology》(影响因子 4.52)的研究为例,阐述生物大数据挖掘在宫颈癌预后相关基因筛选中的应用思路,该研究整合 GEO、TCGA 等多数据库资源,通过多维度分析鉴定出关键预后基因及风险模型。
研究的核心数据来源于 GEO 数据库中的宫颈癌基因芯片数据集 GSE63514,包含 28 例宫颈癌组织样本和 24 例正常宫颈组织样本的基因表达谱数据。这一数据集为差异基因筛选提供了基础,结合 TCGA 数据库的临床信息与甲基化数据,形成了从基因表达差异到功能验证的完整分析链条,体现了生物大数据多源整合的研究优势。
二、差异基因筛选与表达模式分析
2.1 差异基因鉴定与可视化
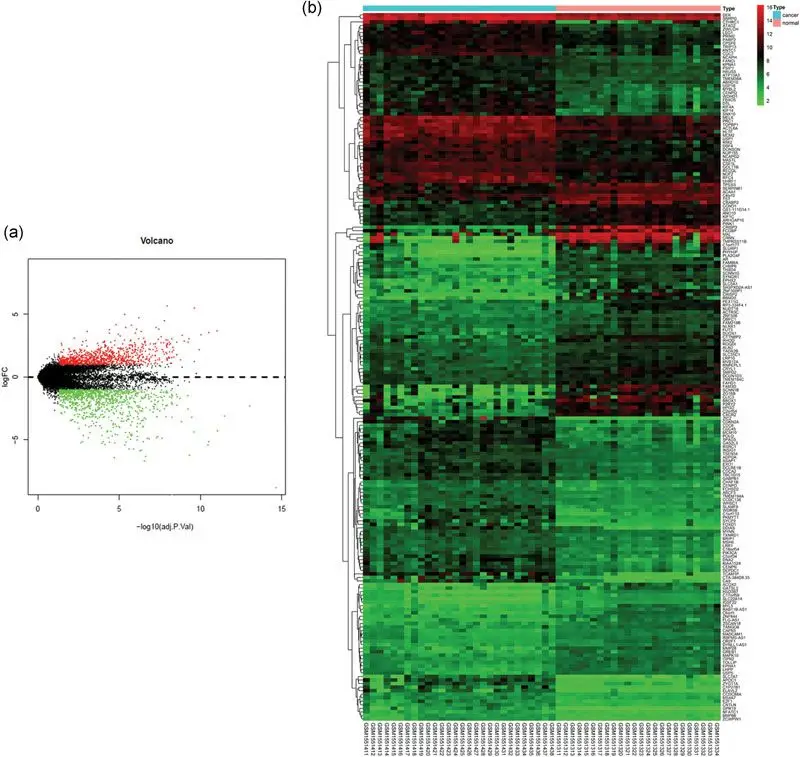
研究首先通过 limma 包对 GSE63514 数据集进行差异表达分析,设定阈值(|log2FC|>1,P<0.05)后共筛选出 1907 个差异基因,其中 944 个基因在宫颈癌组织中上调表达,963 个基因下调表达。为直观展示差异基因分布特征,研究者采用火山图呈现所有差异基因的表达变化趋势,横轴表示表达差异倍数(log2FC),纵轴表示统计学显著性(-log10P),显著上调基因以红色标记,下调基因以蓝色标记,清晰区分了基因表达的整体差异模式。
进一步选取差异最显著的 top200 个基因绘制热图,通过层次聚类分析展示样本间的基因表达异质性。热图中红色表示高表达,绿色表示低表达,结果显示宫颈癌样本与正常样本在基因表达模式上呈现明显聚类,表明差异基因能够有效区分肿瘤与正常组织,为后续功能分析奠定了基础。这一过程充分体现了生物大数据挖掘整合 GEO-TCGA 筛选宫颈癌差异基因的核心逻辑 —— 通过多数据库数据互补,提升差异基因筛选的准确性与可靠性。
2.2 功能注释与富集分析
差异基因的功能注释是解析其生物学意义的关键步骤。研究者将筛选出的差异基因提交至 DAVID 数据库进行 GO(Gene Ontology)功能富集分析和 KEGG(Kyoto Encyclopedia of Genes and Genomes)通路富集分析。GO 分析结果显示,上调基因主要富集于细胞周期调控、DNA 复制、细胞增殖等生物学过程,下调基因则与细胞黏附、免疫应答、激素代谢等功能相关。KEGG 通路分析发现,差异基因显著富集于宫颈癌相关信号通路(如 PI3K-Akt 通路、MAPK 通路)、细胞周期通路及 p53 信号通路,提示这些通路的异常激活或抑制可能参与宫颈癌的发生发展。
三、蛋白互作网络与共表达网络分析
3.1 PPI 网络构建与核心模块筛选
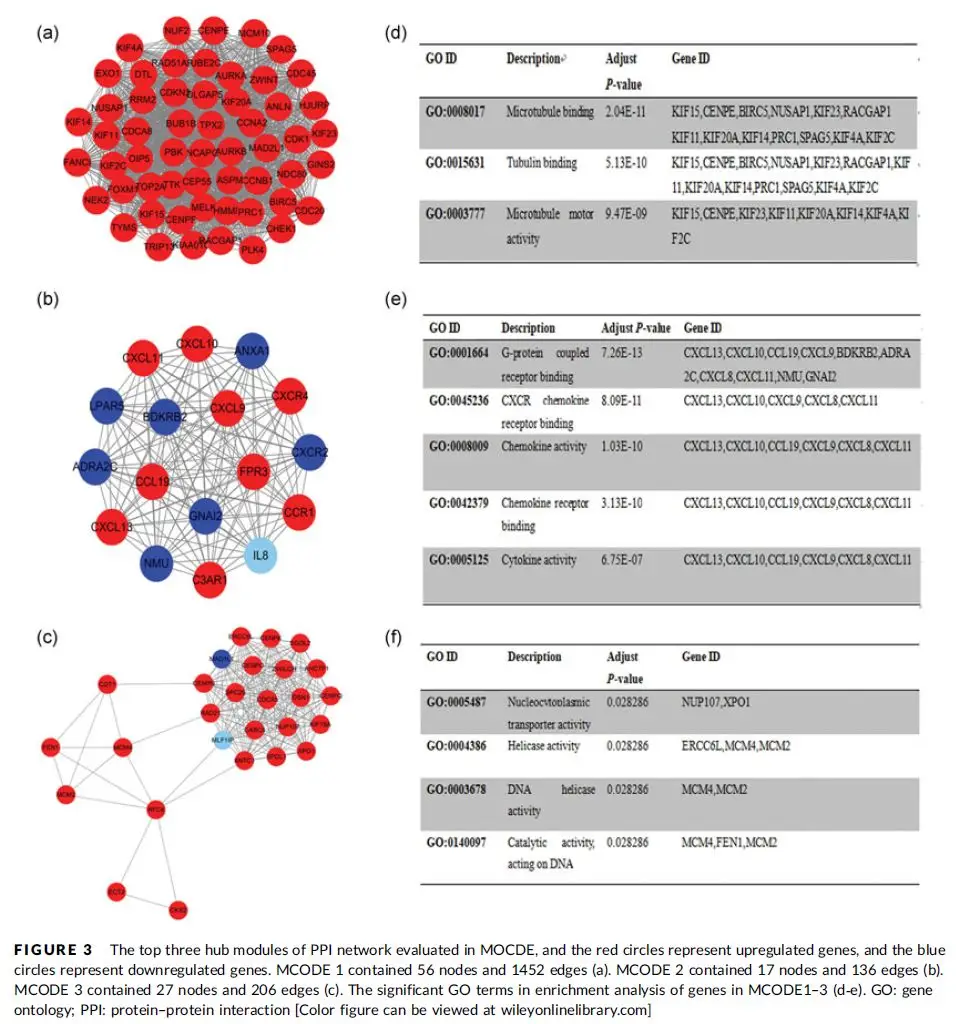
为探索差异基因间的相互作用关系,研究者将差异基因导入 STRING 数据库(置信度阈值 > 0.4),构建蛋白质 - 蛋白质相互作用(PPI)网络,共获得包含 1243 个节点和 3856 条边的互作网络。将网络数据导入 Cytoscape 软件后,利用 MCODE 插件进行模块分析,基于节点连接度、聚类系数等参数筛选出 3 个关键子网络模块。这些子网络中的基因主要参与细胞周期调控、染色体分离、DNA 损伤修复等生物学过程,提示其在宫颈癌恶性增殖中的核心作用。
对核心模块基因的 GO 富集分析进一步验证了上述结果,分子功能层面主要富集于 DNA 结合、蛋白激酶活性、细胞周期调控因子活性等;细胞组分层面集中在细胞核、染色体、纺锤体等结构,表明这些模块基因通过调控细胞分裂过程影响宫颈癌进展。
3.2 WGCNA 共表达网络与关键模块鉴定
利用 R 语言 WGCNA 包对差异基因进行共表达网络分析,通过计算基因表达相关性构建加权共表达网络,采用动态树切割算法将表达模式相似的基因聚类为 4 个共表达模块(以不同颜色标记)。通过模块与表型(宫颈癌 / 正常组织)的相关性分析发现,蓝色模块与宫颈癌呈显著正相关(相关系数 = 0.72,P=4e-09),棕色模块与宫颈癌呈显著负相关(相关系数 =-0.68,P=3e-08)。
模块内基因的连通性分析显示,蓝色模块和棕色模块的基因连通性较高,提示其内部存在紧密的协同表达关系。研究者通过计算基因的模块内连接度(intramodular connectivity)筛选出 116 个 hub 基因(连接度排名前 25%),这些基因作为模块核心调控因子,被认为是参与宫颈癌发生的关键候选基因。上述 PPI 网络与 WGCNA 共表达网络的联合分析,是生物大数据挖掘 PPI-WGCNA 解析宫颈癌分子机制的典型应用,通过多维度网络模型揭示基因间的协同作用及对疾病进程的调控规律。
四、预后相关基因筛选与模型构建
4.1 预后基因的临床验证
将 116 个 hub 基因与 TCGA 数据库中的宫颈癌转录组数据及临床预后信息进行关联分析,通过单因素 Cox 比例风险回归筛选出与总生存期(OS)显著相关的基因(P<0.05),进一步经多因素 Cox 回归分析最终确定 SCNN1B、ANLN、APOC1、CNTLN 和 TEX30 五个基因作为独立预后标志物。基于这五个基因的表达水平和 Cox 回归系数,构建预后风险评分模型:风险评分 =(0.21×SCNN1B 表达)+(0.18×ANLN 表达)+(0.15×APOC1 表达)+(-0.19×CNTLN 表达)+(0.23×TEX30 表达)。
根据风险评分中位数将 TCGA 宫颈癌患者分为高风险组和低风险组,生存分析显示高风险组患者的总生存期显著短于低风险组(HR=2.17,P=0.0003),Kaplan-Meier 生存曲线呈现明显分离。受试者工作特征(ROC)曲线分析显示,该模型预测 5 年生存期的 AUC 值为 0.719,表明其具有较好的预后预测效能。这一成果标志着生物大数据挖掘构建宫颈癌预后风险评分模型的成功实践 —— 通过整合基因表达数据与临床预后信息,实现了从 “基因筛选” 到 “临床实用模型” 的转化。
4.2 甲基化调控基因的筛选
对 hub 基因的进一步挖掘中,GEPIA 数据库生存分析发现 RMI2 和 EPHX2 基因高表达患者的总生存期显著延长(P<0.05)。TCGA 甲基化数据关联分析显示,这两个基因的表达水平与其启动子区甲基化水平呈显著负相关(RMI2:r=-0.38,P=2e-06;EPHX2:r=-0.32,P=3e-04),提示甲基化沉默可能是其在宫颈癌中低表达的重要原因。
生存分析进一步证实,RMI2 基因高甲基化组患者的总生存期显著短于低甲基化组(P=0.023),而 EPHX2 甲基化水平与生存期无显著关联。FireBrowse 和 ONCOMINE 数据库的验证显示,RMI2 和 EPHX2 在多种癌症中均呈现低表达模式,且其低表达与不良预后相关,表明这两个基因可能作为泛癌预后标志物发挥抑癌作用。

五、研究结论与生物大数据应用价值
本研究通过整合 GEO 和 TCGA 等公共数据库资源,采用差异分析、PPI 网络、WGCNA 共表达网络等生物信息学方法,系统筛选出宫颈癌预后相关的关键基因,并构建了具有临床应用价值的预后风险模型。研究亮点在于多数据库交叉验证和多层面分析策略的结合,从基因表达差异到蛋白互作,再到临床预后关联,层层递进揭示了 SCNN1B、ANLN、RMI2 等基因在宫颈癌进展中的作用机制。
生物大数据挖掘技术的应用显著提升了研究效率,通过整合公共数据资源避免了重复实验,降低了研究成本,同时多数据库验证增强了结果的可靠性。此类研究为宫颈癌的早期诊断、预后评估及靶向治疗提供了潜在分子标志物,也为其他恶性肿瘤的生物信息学研究提供了可借鉴的分析框架,凸显了生物大数据在转化医学研究中的重要价值。
乐备实是国内专注于提供高质量蛋白检测以及组学分析服务的实验服务专家,自2018年成立以来,乐备实不断寻求突破,公司的服务技术平台已扩展到单细胞测序、空间多组学、流式检测、超敏电化学发光、Luminex多因子检测、抗体芯片、PCR Array、ELISA、Elispot、多色免疫组化等30多个,建立起了一套涵盖基因、蛋白、细胞以及组织水平实验的完整检测体系。
我们可提供从样本运输、储存管理、样本制备、样本检测到检测数据分析的全流程服务。凭借严格的实验室管理流程、标准化实验室操作、原始数据储存体系以及实验项目管理系统,已经为超过3000家客户单位提供服务,年检测样本超过100万,受到了广大客户的信任与支持。







 沪公网安备31011502400759号
沪公网安备31011502400759号
 营业执照(三证合一)
营业执照(三证合一)




