













一、自噬在疾病发生发展中的关键作用与研究进展
自噬(Autophagy)是细胞通过自我降解与循环利用胞内组分,维持稳态的重要过程。根据作用机制与底物递送途径的不同,自噬主要可分为巨自噬、微自噬和分子伴侣介导的自噬三种类型。作为一种高度保守的防御与应激调控机制,自噬既承担细胞内代谢废物清除与成分回收的功能,又参与抵抗病原体入侵及减轻内源性毒性损伤,在维持细胞正常生理活动中发挥着双重作用。
近年研究显示,自噬功能异常与多种重大疾病的发病机制密切相关,尤其在肿瘤发生发展、神经退行性病变及免疫相关疾病等领域受到广泛关注。其在感染、炎症、心血管疾病等病理过程中的作用亦日益明确,已成为基础与临床研究的重要方向。为进一步揭示自噬的调控网络及其在疾病中的具体角色,学术界持续开展深入探索,积累了丰富的研究成果。以下整理部分相关文献,旨在梳理该领域的研究思路,以期为科研工作提供参考。
二、靶向溶酶体酸化:恢复自噬功能治疗非酒精性脂肪肝病的潜在策略
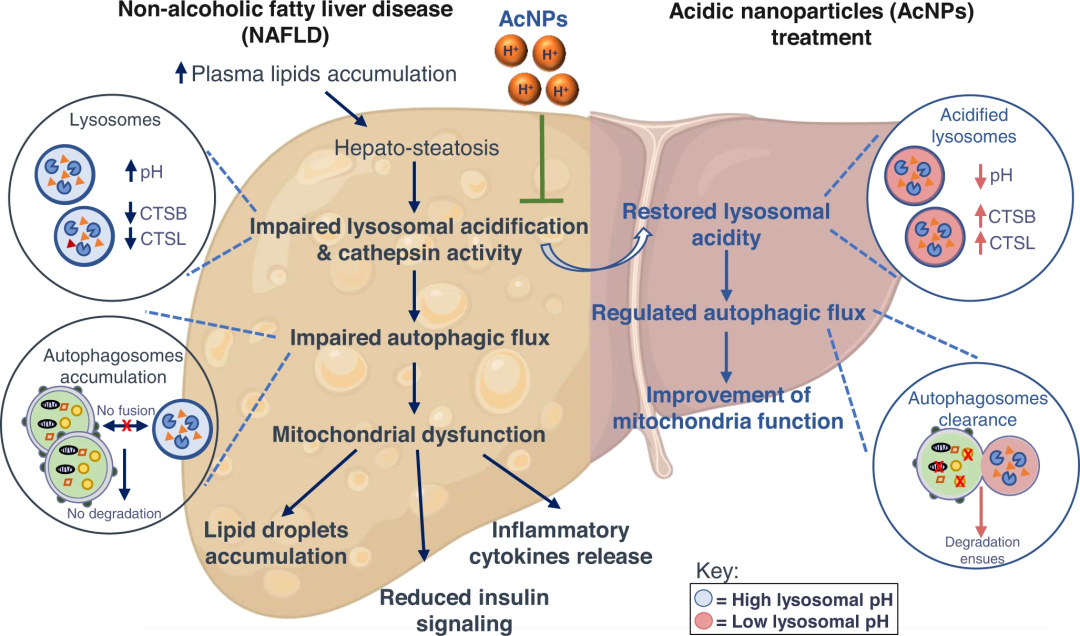
非酒精性脂肪肝病(NAFLD)是全球范围内最为普遍的肝脏疾病之一。研究表明,肝脏内游离脂肪酸水平升高可损害溶酶体酸化过程,进而抑制自噬通量。为探究恢复溶酶体功能能否改善自噬、线粒体功能及胰岛素敏感性,一项发表于《自然·通讯》(IF: 16.6)的研究开发了一类新型可生物降解的酸激活纳米粒子(acNPs),旨在通过靶向调节溶酶体pH值,恢复其正常酸化状态与自噬活性。
该纳米粒子由氟化聚酯构成,在血浆中性pH环境中保持稳定,进入细胞经内吞途径抵达溶酶体后,于酸性环境(pH≈6)中被激活并降解,进一步促进溶酶体酸化,从而增强其功能。在高脂饮食诱导的NAFLD小鼠模型中,acNPs治疗可有效恢复溶酶体酸度,使自噬通量与线粒体功能回归至正常健康水平。与此同时,研究观察到小鼠空腹高血糖及肝脏脂肪变性均得到显著逆转,表明acNPs在NAFLD治疗中具有重要的转化潜力。
本研究提出了一种通过纳米材料靶向恢复溶酶体酸度以调控自噬的代谢干预新途径,为NAFLD的药物研发提供了新的思路。
原文链接:https://www.nature.com/articles/s41467-023-38165-6
三、抑制TRABID通过调控有丝分裂与自噬激活cGAS/STING通路增强抗肿瘤免疫
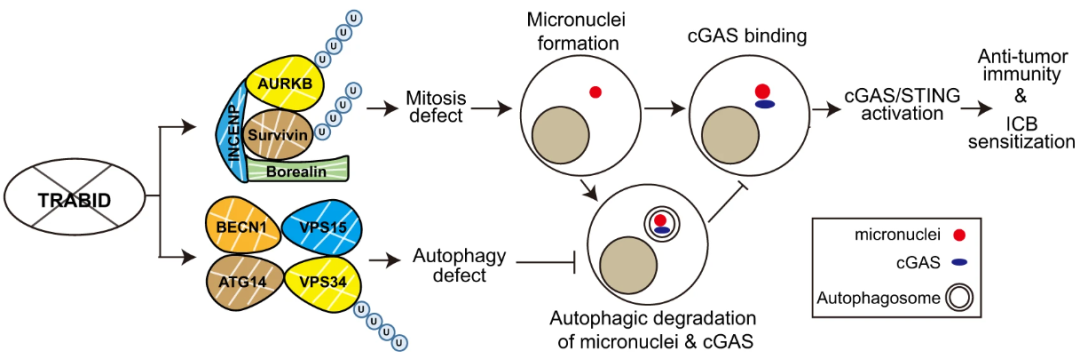
激活肿瘤内在免疫是提升免疫治疗疗效的重要策略。既往研究报道,去泛素化酶TRABID具有促进自噬的作用。本研究发表于《自然·通讯》(IF:16.6),进一步揭示TRABID在抑制抗肿瘤免疫中发挥关键功能。
机制研究表明,TRABID在有丝分裂过程中表达上调,并通过去除Aurora B和Survivin蛋白上的K29连接型多泛素链,维持染色体乘客复合物的稳定性,从而调控有丝分裂进程。抑制TRABID会导致有丝分裂与自噬协调作用失调,引起微核形成,同时使cGAS蛋白免受自噬性降解,进而激活cGAS/STING天然免疫通路。
通过遗传学或药理学手段抑制TRABID,可在临床前小鼠模型中增强抗肿瘤免疫监视,并提升对抗PD-1治疗的敏感性。临床数据分析进一步显示,在多数实体肿瘤类型中,TRABID表达与干扰素相关基因特征及抗肿瘤免疫细胞浸润呈负相关。
本研究系统阐明了TRABID通过协同调控有丝分裂与自噬抑制肿瘤免疫的分子机制,提示TRABID是增强实体肿瘤免疫治疗敏感性的潜在靶点。
原文链接:https://www.nature.com/articles/s41467-023-38784-z
四、AQP4通过抑制Nav1.6介导的星形胶质细胞自噬加剧脓毒症相关脑病认知功能障碍
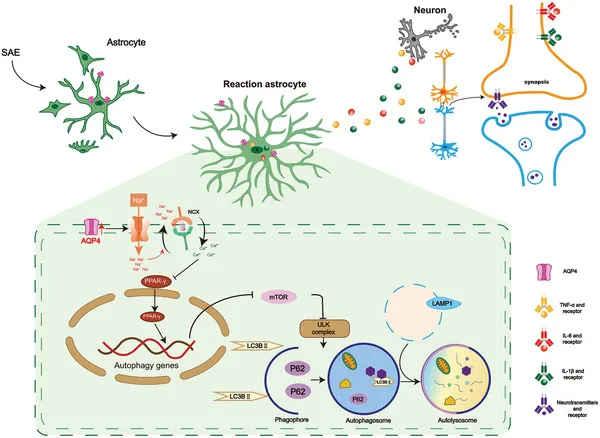
脓毒症相关脑病(SAE)的发病机制与AQP4蛋白介导的星形胶质细胞炎症反应密切相关。本研究发表于《先进科学》(IF:15.1),旨在探究AQP4在SAE中的作用及其导致认知障碍的分子机制。
体内实验结果显示,SAE患者外周血及盲肠结扎穿孔(CLP)模型小鼠皮层与海马组织中AQP4表达均显著上调。进一步研究发现,AQP4缺失可改善CLP或脂多糖(LPS)诱导的学习与记忆功能损伤,其机制与激活星形胶质细胞自噬、抑制星形胶质细胞异常活化及下调促炎细胞因子表达有关。
在机制层面,AQP4敲除能够逆转CLP或LPS引起的星形胶质细胞内PPAR-γ信号抑制,该过程与细胞内钙离子水平及钠通道活性相关。研究表明,AQP4通过下调星形胶质细胞Nav1.6通道,减少胞内钙离子累积,进而抑制PPAR-γ通路,最终阻碍自噬激活并加剧神经炎症。在SAE小鼠模型中,激活自噬、抑制炎症或特异性下调星形胶质细胞Nav1.6表达,均可显著改善AQP4缺失所导致的认知功能损害。
综上,本研究阐明AQP4通过调控Nav1.6-钙信号-PPARγ轴抑制星形胶质细胞自噬,从而加重SAE相关认知障碍,为SAE的病理机制研究与潜在治疗靶点提供了新依据。
原文链接:https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10190498/
五、异银杏素通过靶向CDK6抑制SLC2A1/GLUT1转录诱导AMPK-ULK1介导的细胞毒性自噬抑制肝癌进展
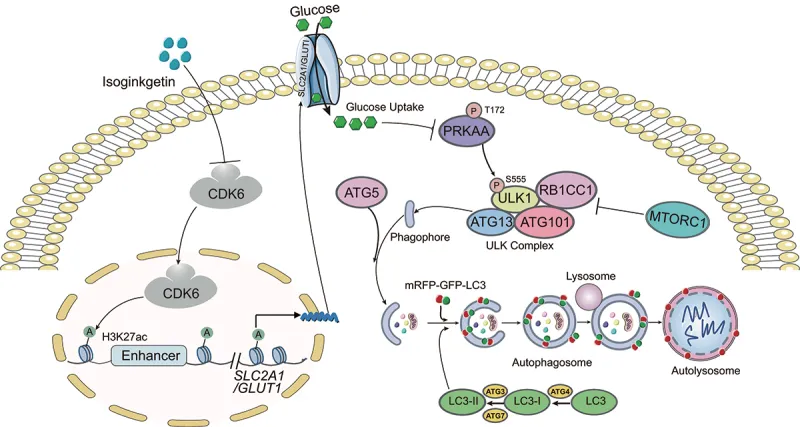
异银杏素(ISO)在多种癌细胞中表现出细胞毒性,但其在肝细胞癌(HCC)中的作用机制尚不明确。本研究发表于《自噬》(IF:13.3),系统揭示了ISO通过靶向CDK6抑制葡萄糖转运、激活自噬通路从而抑制HCC的分子机制。
研究显示,ISO在体外可有效抑制HCC细胞增殖与迁移,并显著诱导LC3-II表达及自噬体形成。使用自噬抑制剂氯喹或敲除自噬关键基因(ATG5或ULK1)可减轻ISO诱导的细胞死亡,表明其作用依赖于细胞毒性自噬的激活。进一步机制研究发现,ISO能显著下调SLC2A1/GLUT1的表达并抑制葡萄糖摄取,进而激活AMPK-ULK1信号通路。过表达SLC2A1/GLUT1可逆转ISO诱导的自噬。
通过分子对接与热稳定性分析,研究证实ISO直接结合于CDK6蛋白的N端结构域并促进其降解。过表达CDK6可消除ISO对SLC2A1/GLUT1转录的抑制及其诱导的自噬效应。表观遗传学分析表明,ISO处理可降低HepG2细胞中SLC2A1/GLUT1增强子区域的H3K27ac、H4K8ac及H3K4me1修饰水平,提示其通过调控组蛋白修饰抑制基因转录。
体内实验证实,ISO能显著抑制HepG2异种移植瘤及化学诱导的原发性HCC小鼠的肿瘤生长。结合TCGA数据及临床组织分析,SLC2A1/GLUT1与CDK6被确认为HCC中具有潜在致癌作用的基因。
本研究阐明ISO通过靶向降解CDK6、抑制SLC2A1/GLUT1转录、降低葡萄糖代谢并激活AMPK-ULK1通路,进而诱导细胞毒性自噬,为HCC的治疗提供了新的潜在策略与作用靶点。
原文链接:https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10012924/
六、总结与展望
自噬作为细胞稳态调控的核心机制,在生理与病理过程中均扮演着复杂而关键的角色。本文梳理了近期在肿瘤、代谢性疾病、神经炎症及免疫调节等领域的重要研究进展,揭示了自噬调控网络的多样性与疾病特异性,并展现了靶向自噬通路在疾病干预中的广阔前景。








 沪公网安备31011502400759号
沪公网安备31011502400759号
 营业执照(三证合一)
营业执照(三证合一)




