







二、小分子降解剂:拓展靶向治疗边界的技术突破
在人类蛋白质组约两万个蛋白质中,据估计约80%因结构或功能特性难以被传统小分子药物干预,通常被归类为“不可成药”靶点。其主要挑战可归纳为以下三方面:
首先,占位驱动型药理学模式对非酶类功能蛋白(如转录因子)作用有限,因其活性不依赖于可被小分子占据的催化位点。其次,部分关键蛋白(如KRAS)表面缺乏深层、明确的疏水性结合口袋,导致小分子难以形成高亲和力、稳定的相互作用。再者,许多通过蛋白质-蛋白质相互作用(PPI)发挥功能的靶点,其结合界面往往宽广且相对平坦,不易被通常较小的小分子化合物有效竞争或阻断。
此外,即使对于已成功开发的靶向药物(如多种EGFR酪氨酸激酶抑制剂),临床应用中亦常出现获得性耐药突变,导致治疗失效,这进一步凸显了开发新作用机制药物的紧迫性。
近年来,以蛋白降解靶向嵌合体(PROTACs)为代表的小分子降解剂技术的发展,为上述困境提供了极具前景的解决方案。该技术通过诱导目标蛋白的降解而非单纯抑制,理论上能够克服由靶点结构特征带来的结合难题,并可能规避因点突变导致的耐药问题,从而为靶向传统“不可成药”蛋白及应对耐药突变开辟了新途径。
三、PROTAC技术:基于事件驱动的靶向蛋白降解新策略
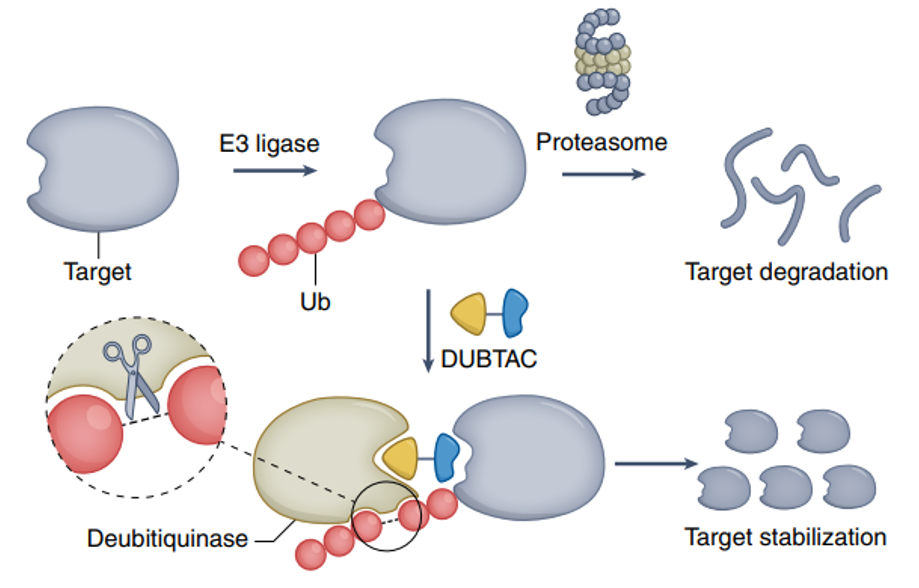
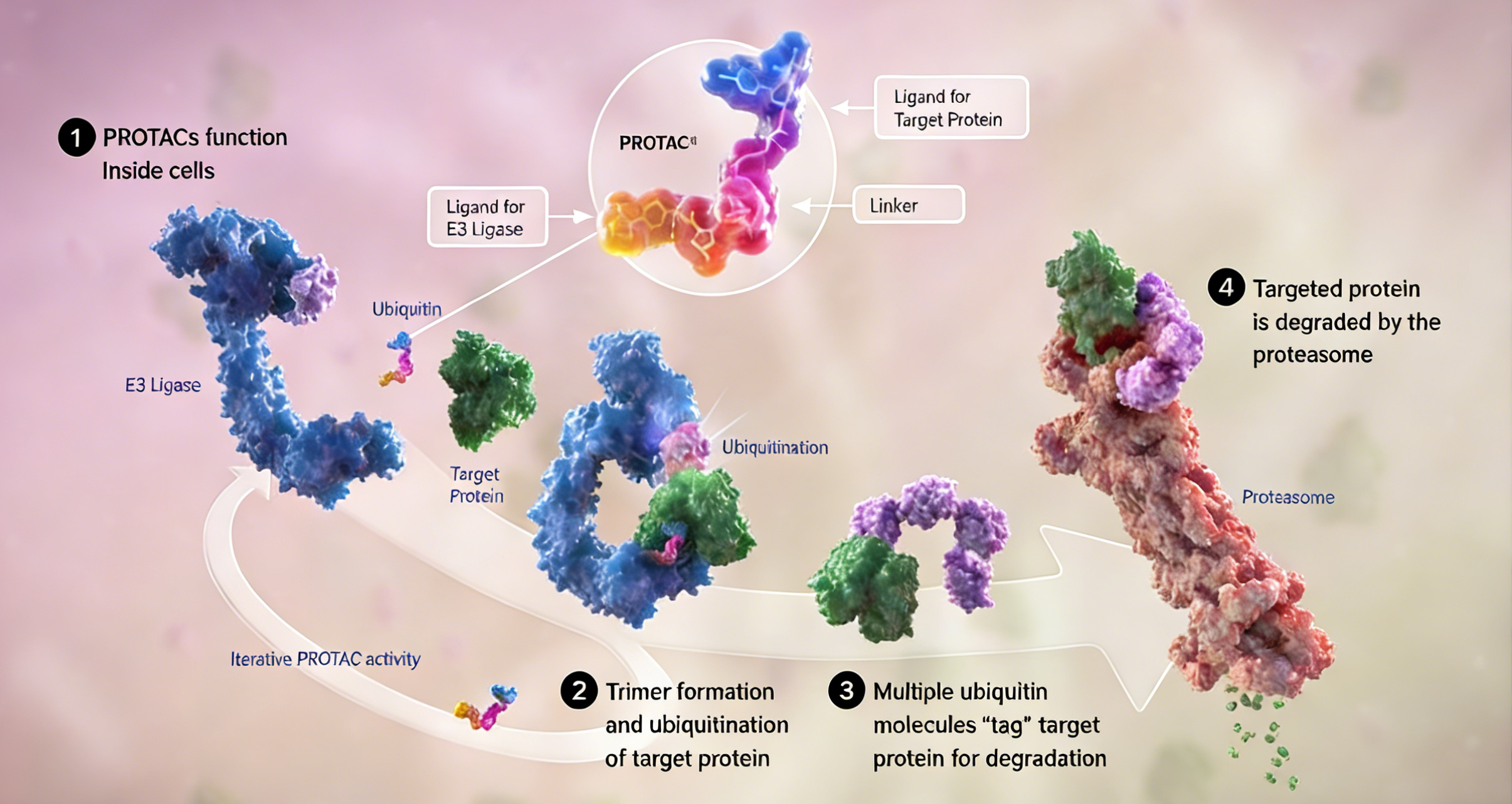
蛋白降解靶向嵌合体(PROTAC)技术是一种利用双功能小分子激活细胞内泛素-蛋白酶体系统,实现对特定目标蛋白选择性降解的前沿策略。该系统的核心作用机制可分为三个关键步骤:首先,E3泛素连接酶识别并催化目标蛋白的单泛素化;随后,通过多轮泛素化反应形成多聚泛素链;最终,带有泛素链标记的靶蛋白被蛋白酶体识别并降解为短肽。
PROTAC分子结构上包含两个功能结构域:其一为靶蛋白结合配体,可特异性识别并结合需降解的目标蛋白;其二为E3泛素连接酶配体,用于招募特定的E3连接酶。两者通过化学连接链共价连接,形成三元复合物,从而将目标蛋白与E3连接酶在空间上拉近,促进靶蛋白的高效泛素化与后续降解。
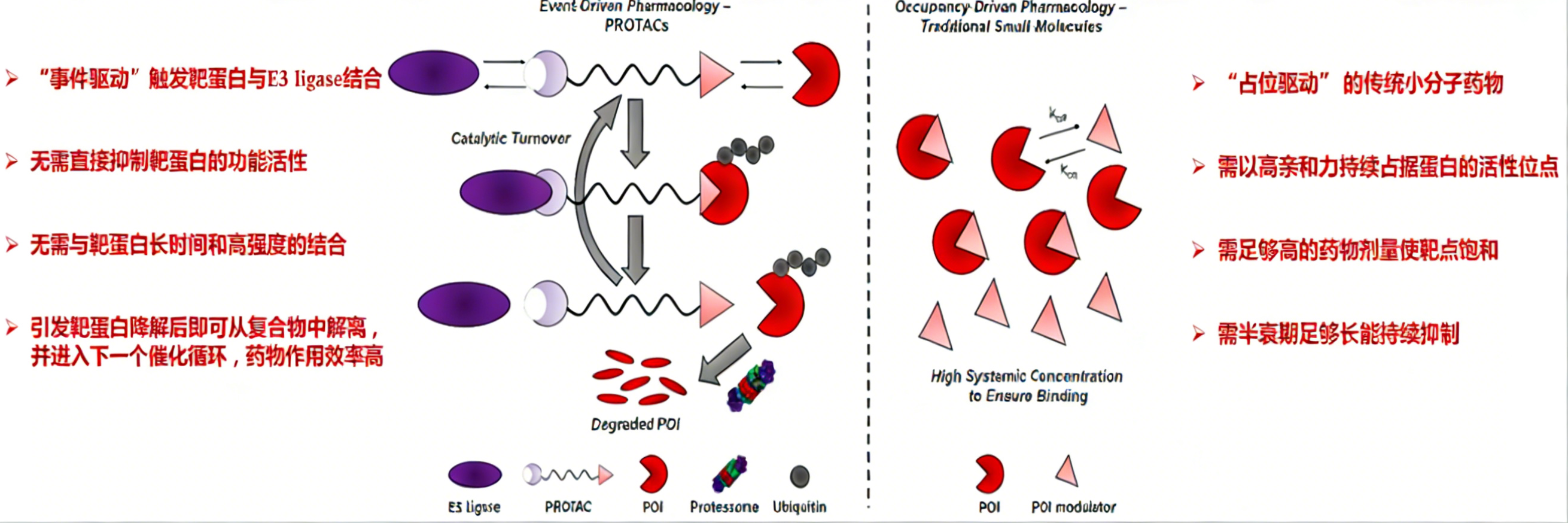
该技术概念于二十一世纪初被提出,其作用机制具有显著创新性。与传统药物的“占位驱动”模式不同——即药物需持续占据靶蛋白活性位点以抑制其功能——PROTAC技术属于“事件驱动”模式。其核心在于催化形成三元复合物,促使靶蛋白泛素化,而无需长期占据靶点或直接抑制其活性。这一特性使其能够靶向缺乏明确活性口袋或结合界面平坦的蛋白质,并可作用于传统小分子或抗体难以干预的靶点。因此,PROTAC技术在理论上具备高选择性、强效降解能力及解决耐药潜力等优势,为拓展药物发现边界提供了新的分子工具。
四、PROTAC技术应对靶点突变耐药的机制与潜力:以BTK为例
以布鲁顿酪氨酸激酶(BTK)为例,其第481位半胱氨酸至丝氨酸的突变(C481S)是导致部分共价抑制剂耐药的主要分子机制之一。该突变破坏了药物与靶点间关键的共价键结合能力,从而使传统抑制剂失效。在此情况下,合理设计的蛋白降解靶向嵌合体(PROTAC)分子可通过同时结合突变型BTK与E3泛素连接酶,诱导靶蛋白发生泛素化并降解,从而绕过突变对抑制剂结合的直接影响。
临床前研究显示,针对BTK开发的PROTAC降解剂能够有效降低野生型及C481S突变型BTK的蛋白水平,并在相应的耐药细胞模型中表现出显著的抗增殖活性。从作用机制而言,由于PROTAC介导的降解不依赖于与特定氨基酸残基形成共价键,因此,只要突变不显著影响PROTAC分子中靶蛋白配体的结合,同一PROTAC分子便有望实现对同一靶点多种不同突变形式的降解。
这一特性为克服由靶点突变引起的获得性耐药提供了新的策略路径,拓展了靶向治疗在应对肿瘤演化与耐药挑战中的应用前景。
五、PROTAC技术的核心优势:事件驱动作用机制的突破
相较于传统小分子药物,PROTAC技术具备独特的靶向蛋白范围扩展能力。其根本区别在于药理学作用机制的差异:传统抑制剂依赖“占位驱动”,而PROTAC则基于“事件驱动”。
传统占位驱动模式的局限性在于,其要求靶蛋白必须具备适合小分子结合的明确活性口袋。抑制剂需以高亲和力持续占据该位点,有效竞争内源性底物,从而阻断蛋白功能。为实现持续抑制,药物需具备足够长的半衰期与较高给药剂量。然而,在已明确的疾病相关蛋白中,约80%的靶点因表面相对平坦、缺乏稳定结合口袋,难以被此类模式有效干预。
与之相对,PROTAC技术通过其双功能分子结构,将靶蛋白与E3泛素连接酶拉近,催化泛素化标记这一“事件”,进而启动蛋白酶体降解途径。该机制不要求药物直接抑制靶蛋白活性,也无需与靶点形成长时间、高强度的结合。理论上,只要靶蛋白表面存在可供瞬时结合的微小结构特征,即可启动降解过程。此外,PROTAC分子在完成一轮催化后能够循环利用,从而以亚化学计量方式实现高效、持续的蛋白清除,展现出显著的作用效率优势。
综上所述,PROTAC技术通过事件驱动的作用范式,突破了传统占位驱动药物的靶点选择性限制,为靶向以往难以成药的蛋白靶点提供了新的可行路径。








 沪公网安备31011502400759号
沪公网安备31011502400759号
 营业执照(三证合一)
营业执照(三证合一)




