







【文献速递】
2026年3月4日,苏州大学吴华、郭凌川教授团队联合苏州系统医学研究所李贵登研究员,在国际顶级代谢期刊 《Nature Metabolism》 (IF=20.8)上发表了一项突破性研究成果。该研究题为 “Extracellular CD44 lactylation impairs CD8+ T cell function in KRAS-mutant colorectal cancer” ,首次揭示了KRAS突变结直肠癌通过一种全新的细胞外机制——诱导CD8+ T细胞表面的CD44蛋白发生乳酸化修饰,从而抑制抗肿瘤免疫,导致免疫治疗耐药。这一发现为克服KRAS突变结直肠癌的免疫治疗抵抗提供了全新的潜在靶点。

一、 研究背景:KRAS突变驱动的免疫逃逸迷雾
结直肠癌是全球常见的恶性肿瘤之一,其中携带KRAS基因突变的亚型(KRAS-MT CRC)尤为棘手。这类患者约占结直肠癌病例的大多数,且通常属于微卫星稳定型,对以PD-1/PD-L1抗体为代表的免疫检查点抑制剂疗法响应率极低。
此前研究已知,KRAS突变是肿瘤免疫逃逸的重要推手,但其背后复杂的分子机制尚不完全清楚。特别是,KRAS突变如何重塑肿瘤微环境,进而抑制关键抗癌免疫细胞——CD8+ T细胞的功能,一直是领域内的研究热点和难点。本研究正是为了揭开这层迷雾。
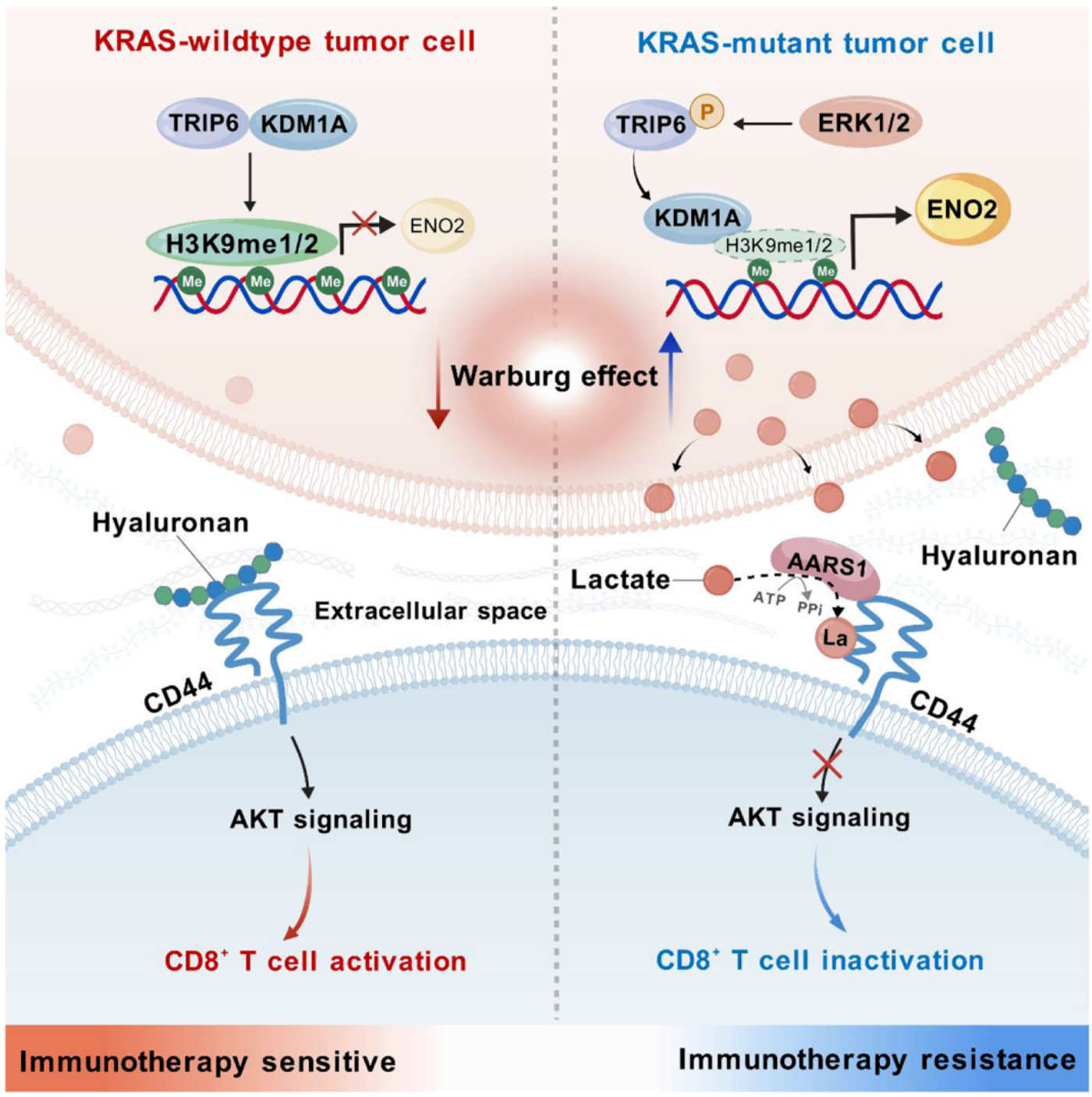
TRIP6在调节CRC中的双重作用
Yang Y, Wu Y,et al. Extracellular CD44 lactylation impairs CD8+ T cell function in KRAS-mutant colorectal cancer. Nat Metab. 2026 Mar 4.
二、 核心发现:一条连接代谢与免疫的“乳酸化信号轴”
研究团队通过一系列精密的分子生物学和细胞生物学实验,逐步揭示了从KRAS突变到T细胞功能抑制的完整信号通路。
关键机制解读:
(1)上游开关:TRIP6的磷酸化
在KRAS突变的结直肠癌细胞中,过度活跃的ERK1/2激酶会磷酸化一个名为TRIP6的蛋白。这一磷酸化事件,是整条信号通路的启动开关。
(2)核心代谢环节:解除抑制,激活糖酵解
正常情况下,未磷酸化的TRIP6会与一个叫做KDM1A的蛋白结合,共同抑制糖酵解关键酶——烯醇酶2 (ENO2) 的表达。但当TRIP6被磷酸化后,它与KDM1A的结合被破坏,对ENO2的抑制随之解除。ENO2表达上调,极大地增强了肿瘤细胞的糖酵解通量,导致大量乳酸产生并分泌到细胞外,形成免疫抑制性的高乳酸微环境。
(3)全新发现:细胞外的“乳酸化阴谋”
传统观点认为乳酸主要在细胞内作为信号分子或代谢燃料。但本研究发现,升高的细胞外乳酸能直接作用于肿瘤浸润的CD8+ T细胞,诱导其表面的CD44蛋白发生一种全新的翻译后修饰——乳酸化。
(4)功能损害与免疫逃逸
CD44是T细胞表面一个重要的黏附与共刺激分子,其功能依赖于与配体透明质酸的结合。研究发现,CD44的乳酸化修饰损害了其与透明质酸的结合能力,进而削弱了T细胞内部的AKT信号通路活性。最终,CD8+ T细胞的增殖、活化和杀伤功能被严重抑制,肿瘤成功实现了免疫逃逸。
三、 转化意义:靶向新轴,破解免疫治疗耐药
基于上述发现,研究团队进一步探索了其临床转化潜力。他们在临床前模型中证实,使用一种专门设计的小鼠TRIP6肽,可以有效阻断TRIP6的磷酸化。
恢复T细胞功能:阻断TRIP6磷酸化,相当于关掉了整条信号轴的“开关”,能够有效恢复CD8+ T细胞的抗肿瘤功能。
增敏免疫疗法:更重要的是,这种策略显著改善了肿瘤对抗PD-1治疗的响应。这意味着,联合靶向TRIP6–ENO2–CD44乳酸化信号轴,可能是克服KRAS突变结直肠癌免疫治疗耐药性的一种极具前景的新策略。
四、 总结与展望
该研究具有多重重要意义:
(1)机制创新:首次发现并阐明了细胞外蛋白(CD44)的乳酸化修饰作为一种全新的免疫逃逸机制,极大地拓展了人们对乳酸在肿瘤微环境中作用的认识。
(2)靶点新颖:鉴定出TRIP6作为一个连接KRAS致癌信号与代谢重编程、免疫抑制的关键节点,为药物开发提供了全新靶点。
(3)临床价值:为占绝大多数的、对现有免疫疗法不敏感的KRAS突变结直肠癌患者,提供了克服耐药的潜在联合治疗策略。
总之,这项发表于《Nature Metabolism》的研究,不仅揭示了KRAS突变结直肠癌免疫逃逸的“新阴谋”,也为未来开发更有效的癌症免疫组合疗法指明了新方向。








 沪公网安备31011502400759号
沪公网安备31011502400759号
 营业执照(三证合一)
营业执照(三证合一)




