







摘要
肝纤维化是慢性肝病进展为肝硬化乃至肝癌的关键步骤,其本质是肝星状细胞(HSC)持续活化导致的细胞外基质过度沉积。细胞焦亡作为一种溶解性、促炎性的程序性细胞死亡方式,近年被证实深度参与肝纤维化的发生与发展。本文系统梳理了细胞焦亡的经典与非经典分子通路,并重点阐述了不同肝脏细胞(包括HSC、肝实质细胞、肝巨噬细胞、肝窦内皮细胞及树突状细胞)焦亡在肝纤维化进程中的具体作用机制。现有研究表明,NOD样受体蛋白3(NLRP3)炎症小体介导的焦亡通路在其中发挥核心作用,其激活可促发级联反应,通过释放白细胞介素(IL)-1β、IL-18等炎症因子,驱动HSC活化并加剧纤维化进程。此外,干扰素激活基因2蛋白(AIM2)、NLRP6等其他炎症小体及多条信号通路亦被证实参与调控。最后,本文探讨了靶向细胞焦亡治疗肝纤维化的潜在策略与未来挑战,以期为该领域的深入研究与临床转化提供参考。
一、细胞焦亡的分子机制概述
细胞焦亡的发生主要由两条信号通路介导:
1. 经典焦亡途径:由炎症小体复合物启动。炎症小体通常由感受蛋白(如NLRP3、AIM2)、衔接蛋白(ASC)和效应蛋白(Pro-Caspase-1)组成。在识别病原相关分子模式(PAMP)或损伤相关分子模式(DAMP)后,炎症小体组装并激活Caspase-1。活化的Caspase-1一方面切割GSDMD蛋白,释放其N端结构域(GSDMD-N)在细胞膜上打孔,另一方面切割IL-1β和IL-18的前体,使其成熟并通过膜孔释放,引发焦亡和炎症级联反应。
2. 非经典焦亡途径:在人源细胞中由Caspase-4/5(鼠源为Caspase-11)介导。这些Caspase可直接识别并结合胞内脂多糖(LPS),被激活后直接切割GSDMD,引发焦亡。此过程同样可间接激活NLRP3炎症小体,促进IL-1β的成熟与释放。
GSDM蛋白家族是焦亡的执行者。除GSDMF外,多数家族成员(如GSDMD、GSDME)均含有自抑制的C端结构域和成孔的N端结构域。Caspase切割释放的N端结构域具有结合膜脂并在膜上寡聚形成孔洞的活性,最终导致细胞渗透压改变而破裂。
二、不同肝脏细胞焦亡在肝纤维化中的作用
(一)肝星状细胞(HSC)焦亡:纤维化的核心驱动
HSC的活化是肝纤维化的核心事件,而HSC自身的焦亡可直接加剧这一过程。
1. NLRP3炎症小体的核心作用:多项研究证实,在HSC中特异性激活NLRP3炎症小体,可直接促进其向肌成纤维细胞转化,并上调α-SMA及I型胶原的表达。例如,醛固酮、甘氨鹅脱氧胆酸等刺激物可通过上调NLRP3表达促进HSC活化,而NLRP3基因敲除或使用其抑制剂(如法尼醇X受体激动剂)则可显著减轻肝纤维化程度。
2. 炎症因子的旁分泌放大效应:HSC发生焦亡后,释放的IL-1β和IL-18可作用于周边未发生焦亡的HSC,通过旁分泌形式进一步激活它们,形成级联放大效应,推动纤维化进展。
3. Ang II的双通路激活机制:血管紧张素II(Ang II)被证实可通过促进活性氧(ROS)生成,同时激活NLRP3/Caspase-1/GSDMD经典通路和Caspase-4/5/GSDMD非经典通路,诱导人HSC-LX2细胞焦亡,并促进IL-1β、IL-18和α-SMA的释放。使用ROS抑制剂或GSDMD抑制剂可阻断此效应,提示了潜在的治疗靶点。
(二)肝实质细胞焦亡:纤维化的起始扳机
肝细胞损伤是慢性肝病的起始事件。肝细胞发生焦亡时,不仅自身死亡,还释放DAMPs和炎症小体,激活邻近的HSC。
1. NLRP3的关键作用:全身性NLRP3敲入小鼠表现出更显著的肝细胞焦亡和肝纤维化。过度激活的肝细胞可将NLRP3炎症小体释放至胞外,被HSC内吞后促进其活化。
2. STING-NLRP3-GSDMD轴:干扰素基因刺激因子(STING)被证实可通过表观遗传调控上调NLRP3表达,从而促进肝细胞焦亡。
3. 保护性分子的发现:生长阻滞特异性转录因子5(GAS5)可通过抑制肝细胞焦亡,间接抑制HSC活化和肝纤维化进程。这些发现为靶向肝细胞焦亡干预纤维化提供了新思路。
(三)肝巨噬细胞焦亡:炎症与纤维化的桥梁
肝巨噬细胞在肝损伤中扮演着炎症反应的核心角色,其焦亡可通过释放大量促炎因子,为HSC活化创造“炎性微环境”。
1. 多条信号通路汇于NLRP3:研究表明,S100A8蛋白通过激活TLR4/NF-κB通路和诱导ROS生成,促进巨噬细胞NLRP3依赖性焦亡。
此外,METTL3/MALAT1/PTBP1/USP8/TAK1通路也被证实可促进巨噬细胞焦亡并向促炎的M1表型极化。
2. 中性粒细胞胞外陷阱(NET)的参与:NET可被肝巨噬细胞吞噬,其携带的DNA激活巨噬细胞内的AIM2炎症小体,诱导焦亡并释放IL-1β/IL-18,进而活化HSC。这一发现连接了中性粒细胞、巨噬细胞和HSC在纤维化中的相互作用。
3. 潜在干预与生物标志物:天然产物熊果酸被发现可通过抑制NOX2介导的NLRP3活化,减少巨噬细胞焦亡而减轻肝纤维化。更有研究提示,血清GSDMD水平可能与肝纤维化分期相关,有望成为新的无创诊断标志物。
(四)其他肝脏细胞的焦亡
1. 树突状细胞(DC):在曼氏血吸虫感染模型中,虫卵抗原可刺激肝脏DC激活NLRP6炎症小体,引发DC焦亡并释放炎症因子,从而活化HSC并促进纤维化。NLRP6敲除小鼠则表现出纤维化减轻。
2. 肝窦内皮细胞(LSEC):在胆汁淤积模型中,NET可激活LSEC中的NLRP3炎症小体,诱发LSEC焦亡,加剧肝内炎症、凝血和纤维化反应。
三、总结与展望
综上所述,细胞焦亡作为一种由炎症小体介导、GSDM蛋白执行的程序性坏死,通过影响肝脏内多种细胞,在肝纤维化进程中扮演了关键驱动角色。HSC是焦亡信号的最终效应器,而肝细胞和巨噬细胞的焦亡则作为上游信号源,通过释放IL-1β/IL-18等介质,构成一个多细胞参与的“焦亡-炎症-活化”网络,共同推动ECM沉积和纤维化进展。
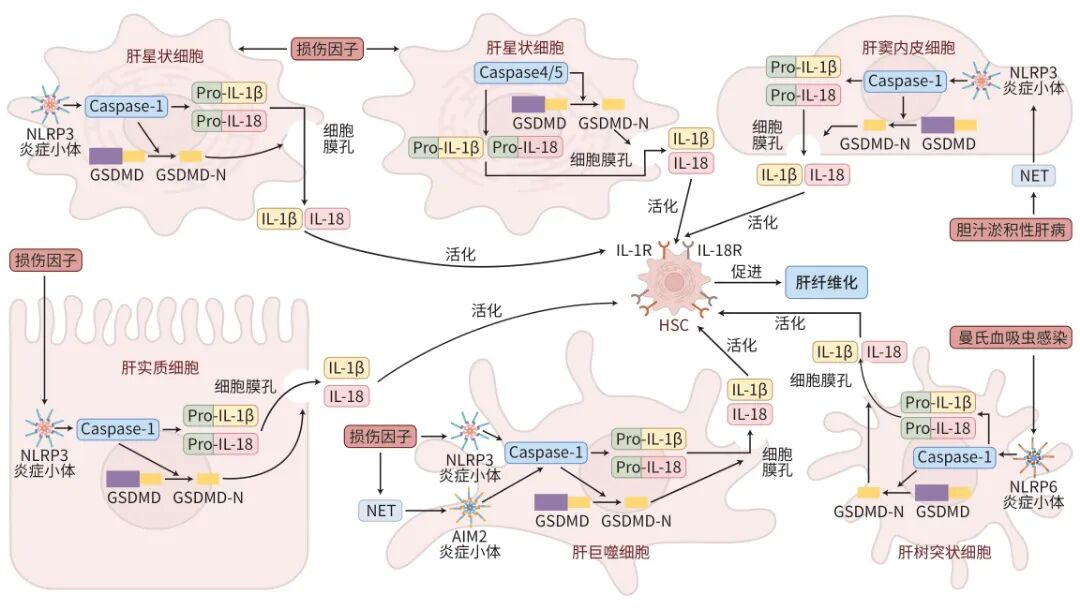
不同肝细胞焦亡促进肝纤维化的机制图
金泳帆, 赵春梅, 邰文琳. 细胞焦亡在肝纤维化中的作用及研究进展[J]. 临床肝胆病杂志, 2026, 42(1): 197-202
当前研究主要聚焦于NLRP3炎症小体,其抑制剂MCC950在动物模型中显示出良好的抗纤维化潜力。此外,靶向AIM2、NLRP6等其他炎症小体,或阻断IL-1β/IL-18与其受体的结合,也是极具前景的策略。
然而,将细胞焦亡调控转化为临床疗法仍面临严峻挑战:
1. 免疫稳态的维持:细胞焦亡是先天免疫的重要组成,广泛抑制炎症小体可能增加感染或肿瘤风险。未来需要开发能精准靶向特定肝脏细胞(如活化HSC)或特定通路(如疾病状态下异常激活的NLRP3)的干预手段。
2. 效应的精准阻断:鉴于炎症因子是激活HSC的直接推手,开发特异性拮抗HSC表面IL-1R或IL-18R的药物,可能是一种更下游、更特异的治疗策略,有望在阻断纤维化信号的同时,保留上游的部分免疫功能。
未来研究需深入探索不同病因、不同阶段肝纤维化中细胞焦亡的精细分子机制,并在此基础上发展安全、精准的干预策略,方能为肝纤维化患者带来新的治疗曙光。








 沪公网安备31011502400759号
沪公网安备31011502400759号
 营业执照(三证合一)
营业执照(三证合一)




