







一、引言
炎症作为机体防御感染、清除损伤组织的基础免疫应答,启动于危险信号识别,经多分子协同传导实现局部防御,完成任务后有序消退并启动组织修复。当刺激持续、信号放大过度或负调控失效时,急性炎症转为慢性失控状态,参与动脉粥样硬化、代谢疾病、自身免疫病、肿瘤等多种疾病进程。 现有研究多聚焦单一分子或通路,缺乏对整体网络的系统性梳理。本文以危险信号识别—胞内传导—基因转录—炎症释放—放大与消退—失衡致病为主线,完整解析炎症信号网络,并提炼科研应用要点,助力机制研究与实验设计。
二、炎症反应的启动:危险信号的识别
(一)危险信号来源
炎症启动的核心是哨兵细胞识别异常信号,主要包括巨噬细胞、树突状细胞、上皮细胞等固有免疫感知细胞。
- PAMPs(病原体相关分子模式):细菌LPS、病毒核酸、真菌细胞壁成分等病原体保守特征。
- DAMPs(损伤相关分子模式):组织损伤、细胞坏死释放的内源性报警信号,如ATP、热休克蛋白、核蛋白片段等。
(二)模式识别受体(PRR)
PRR是细胞感知危险信号的核心受体家族,其中Toll样受体(TLR) 为最经典的固有免疫探测器。
- TLR4:识别革兰氏阴性菌LPS
- TLR3:识别病毒双链RNA
- 功能:识别危险信号并将胞外刺激转化为胞内信号,启动下游级联反应。
三、核心炎症信号通路:TLR/MyD88/NF-κB轴
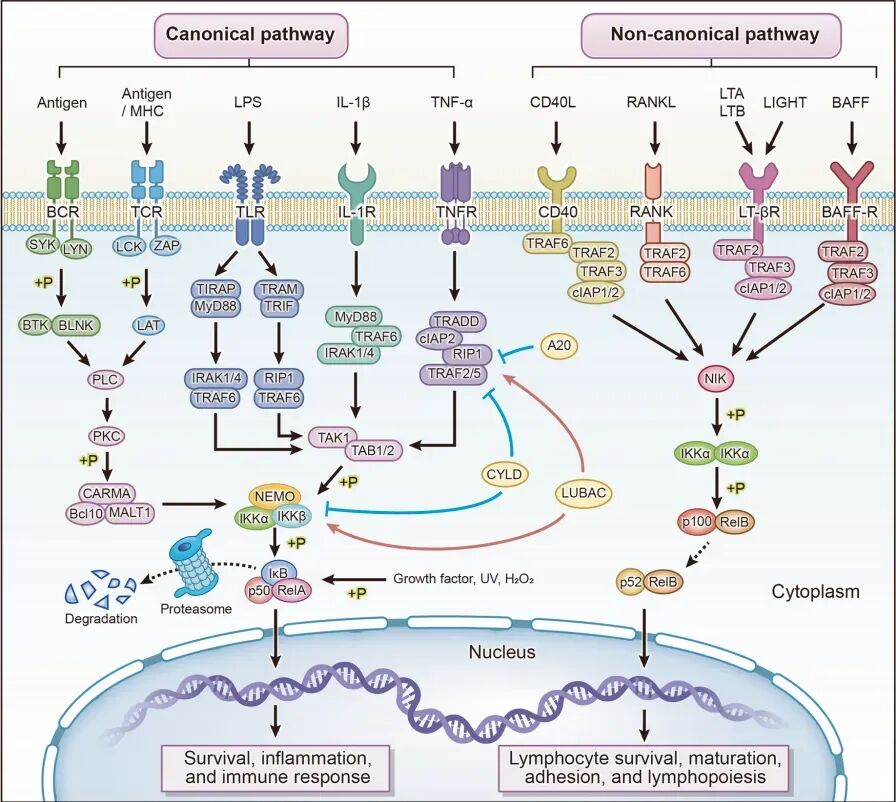
(一)信号中转:MyD88适配蛋白
TLR激活后招募MyD88作为关键信号中转站,衔接受体与下游激酶复合物,形成TLR→MyD88→IRAK→TRAF6→TAK1的传导链。
注:TLR3主要不依赖MyD88,TLR4可并行TRIF通路,经典主线以TLR/MyD88为主。
(二)信号解锁:IKK复合体与IκB降解
TAK1激活IKK复合体,催化抑制蛋白IκB磷酸化并经泛素-蛋白酶体途径降解,解除对NF-κB的胞质锚定抑制。
- IκB:NF-κB的“分子锁”
- IKK:炎症信号的“解锁开关”
(三)基因转录调控:NF-κB转录因子
NF-κB释放后暴露核定位序列,转位入核结合炎症基因启动子,上调TNF-α、IL-6、IL-1β、COX-2、iNOS等关键促炎介质表达,将胞外危险信号转化为转录应答。
(四)炎症扩散:炎症因子的胞外广播
转录合成的炎症因子分泌至胞外,招募中性粒细胞、单核细胞,激活血管内皮,启动局部组织炎症,实现从单细胞反应到多细胞协同防御的转变。
四、炎症的动态调控:放大、消退与失衡
(一)炎症信号的正反馈放大
初始炎症因子激活周围细胞与免疫细胞,进一步释放更多炎症介质,形成信号放大循环,快速增强防御强度;过度放大则走向失控。 危险信号→哨兵细胞激活→炎症因子释放→免疫细胞募集→炎症级联放大
(二)炎症的负调控与消退
正常炎症具备完整“油门-刹车”系统,保障适时终止:
- IκBα重新合成抑制NF-κB
- 负调控蛋白阻断上游TLR信号
- 抗炎因子与修复型巨噬细胞主导消退与组织重建
(三)炎症失控与慢性炎症
失控诱因:持续刺激、放大过度、刹车失效、损伤-炎症恶性循环、修复失败。
慢性炎症特征:长期低强度激活、免疫细胞持续浸润、氧化应激增强、组织重塑异常、纤维化与结构破坏,驱动多系统慢性疾病。
五、炎症信号网络的完整通路图谱
外来病原体/组织损伤→PAMPs/DAMPs→哨兵细胞识别→PRR/TLR激活→MyD88信号中转→IRAK/TRAF6/TAK1级联→IKK激活→IκB降解→NF-κB核转位→炎症基因上调→炎症因子释放→局部炎症→危险清除→消退修复;或刺激持续/调控失效→慢性炎症→疾病发生发展
六、科研应用:机制研究思路与常见误区
(一)通路机制验证的完整证据链
以“药物调控TLR4/MyD88/NF-κB减轻炎症”为例,需分层验证:
1. 上游:TLR4表达/活化变化
2. 中转:MyD88、IRAK、TRAF6等信号分子
3. 核心节点:IKK活性、IκB降解、NF-κB磷酸化与核转位
4. 下游:TNF-α、IL-6、IL-1β等因子水平
5. 表型:组织损伤、免疫浸润、功能指标改善
(二)炎症研究五大常见误区
1. 误将炎症因子升高等同于机制阐明
2. 过度聚焦NF-κB,忽略MAPK、JAK-STAT、炎症小体等通路
3. 仅检测mRNA,忽视蛋白水平与分泌状态
4. 通路变化直接等同于功能改善,缺乏表型证据
5. 追求“无炎症”,忽视炎症的生理防御价值
七、结论与展望
炎症并非单一分子事件,而是危险识别—信号传导—基因表达—细胞动员—组织重塑的动态网络。TLR/MyD88/NF-κB构成核心传导轴,配合正负调控实现防御与修复的平衡;失衡则引发慢性炎症并参与疾病。未来研究应从单一通路转向网络调控,聚焦时空动态、细胞特异性与交叉对话,为精准抗炎治疗提供新靶点与策略。








 沪公网安备31011502400759号
沪公网安备31011502400759号
 营业执照(三证合一)
营业执照(三证合一)




