







摘要
肿瘤患者常表现为全身性缺铁,但肿瘤微环境(TME)因肿瘤坏死呈现局部铁过载,其对CD8+T细胞的影响与机制尚不明确。本研究证实,TME铁过载通过慢性TCR激活与肿瘤来源铁调素(Hepcidin) 双重抑制铁输出蛋白SLC40A1,导致CD8+T细胞内铁蓄积、脂质过氧化升高,最终诱发铁死亡与功能耗竭。恢复SLC40A1表达或使用铁螯合剂可逆转该过程;SLC40A1过表达CAR-T可抵抗铁过载杀伤,显著提升实体瘤治疗效果。本研究揭示铁代谢调控抗肿瘤免疫的新机制,为优化免疫细胞治疗提供工程化新策略。
一、引言
铁代谢参与肿瘤细胞增殖与免疫细胞功能重塑,CD8+T细胞是抗肿瘤免疫核心效应细胞,但其铁稳态调控机制尚未阐明。临床观察到肿瘤患者全身缺铁而肿瘤局部铁富集,补铁干预对肿瘤免疫的影响存在争议。正常淋巴液铁浓度远低于血液,而TME因大量肿瘤细胞坏死形成显著铁过载微环境,这种局部高铁是否损伤CD8+T细胞、通过何种信号通路影响功能、能否用于优化CAR-T治疗,是亟待解决的关键科学问题。
本研究提出科学假说:肿瘤坏死导致TME铁过载;慢性TCR激活与肿瘤铁调素协同下调SLC40A1,造成CD8+T细胞铁蓄积,诱发铁死亡与功能耗竭;恢复SLC40A1或铁螯合可逆转该过程,SLC40A1过表达CAR-T可增强实体瘤疗效。
二、材料与方法
(一)临床样本
收集结直肠癌、肺癌患者外周血、肿瘤间隙液(TIF)、胸水及肿瘤组织;健康供者外周血与淋巴液作为对照。
(二)动物模型
构建小鼠皮下肿瘤模型,分为高铁饮食组、正常饮食组、铁螯合剂(DFP)处理组、铁调素敲除/过表达组。
(三)细胞实验
分离并活化人/小鼠CD8+T细胞,体外给予不同浓度铁处理,联合铁死亡抑制剂(Fer-1、Lip-1)、铁调素干预;构建SLC40A1过表达CAR-T细胞。
(四)检测指标
- 铁浓度:组织/细胞铁含量、普鲁士蓝染色
- 细胞死亡与铁死亡:流式细胞术、脂质过氧化、ROS、线粒体形态
- 功能检测:IFN-γ、TNF-α、IL-2分泌,增殖能力,耗竭标志物表达
- 分子机制:qPCR、Western blot、多核糖体分析、空间转录组
三、结果
(一)TME铁过载与CD8+T细胞耗竭正相关
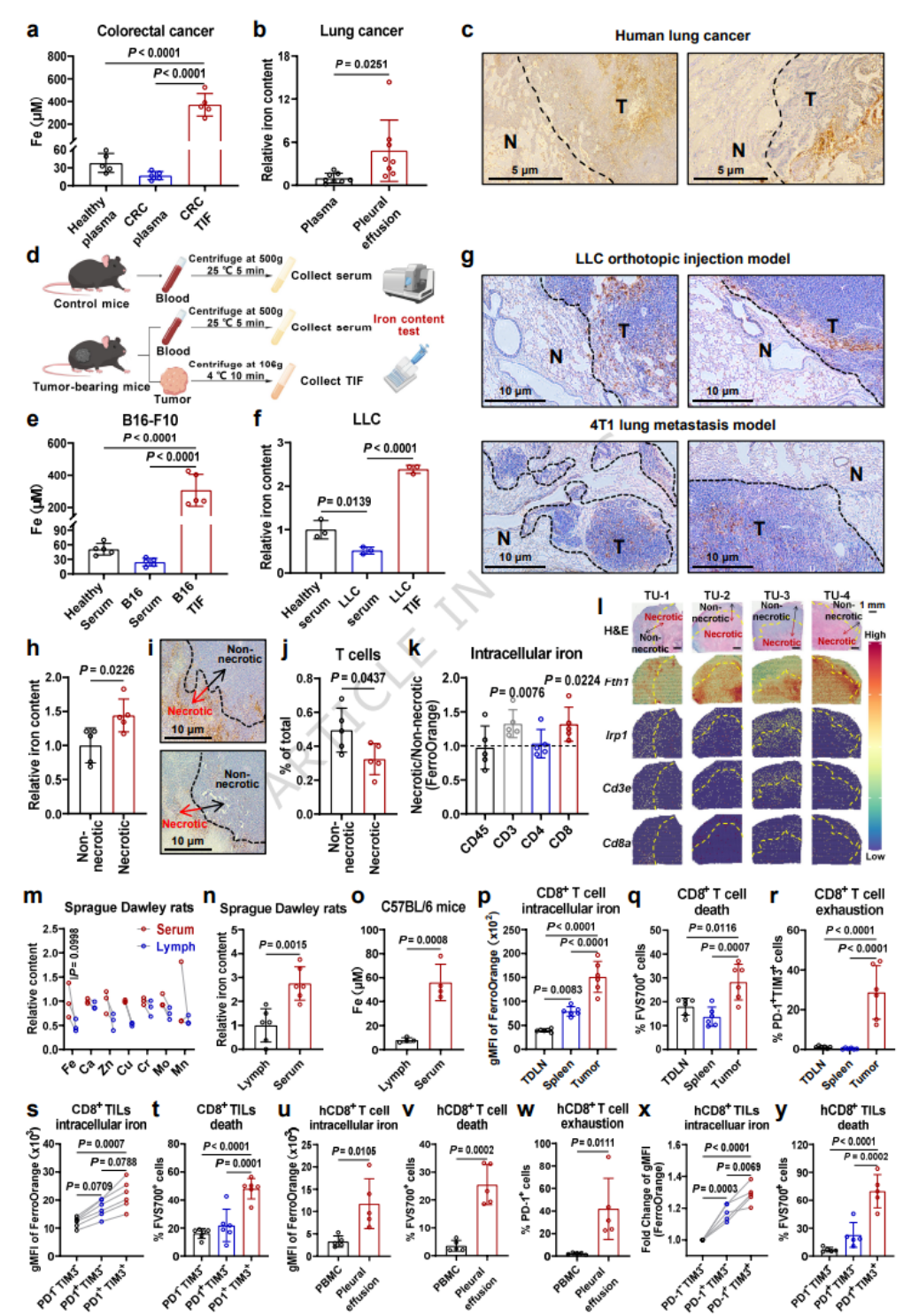
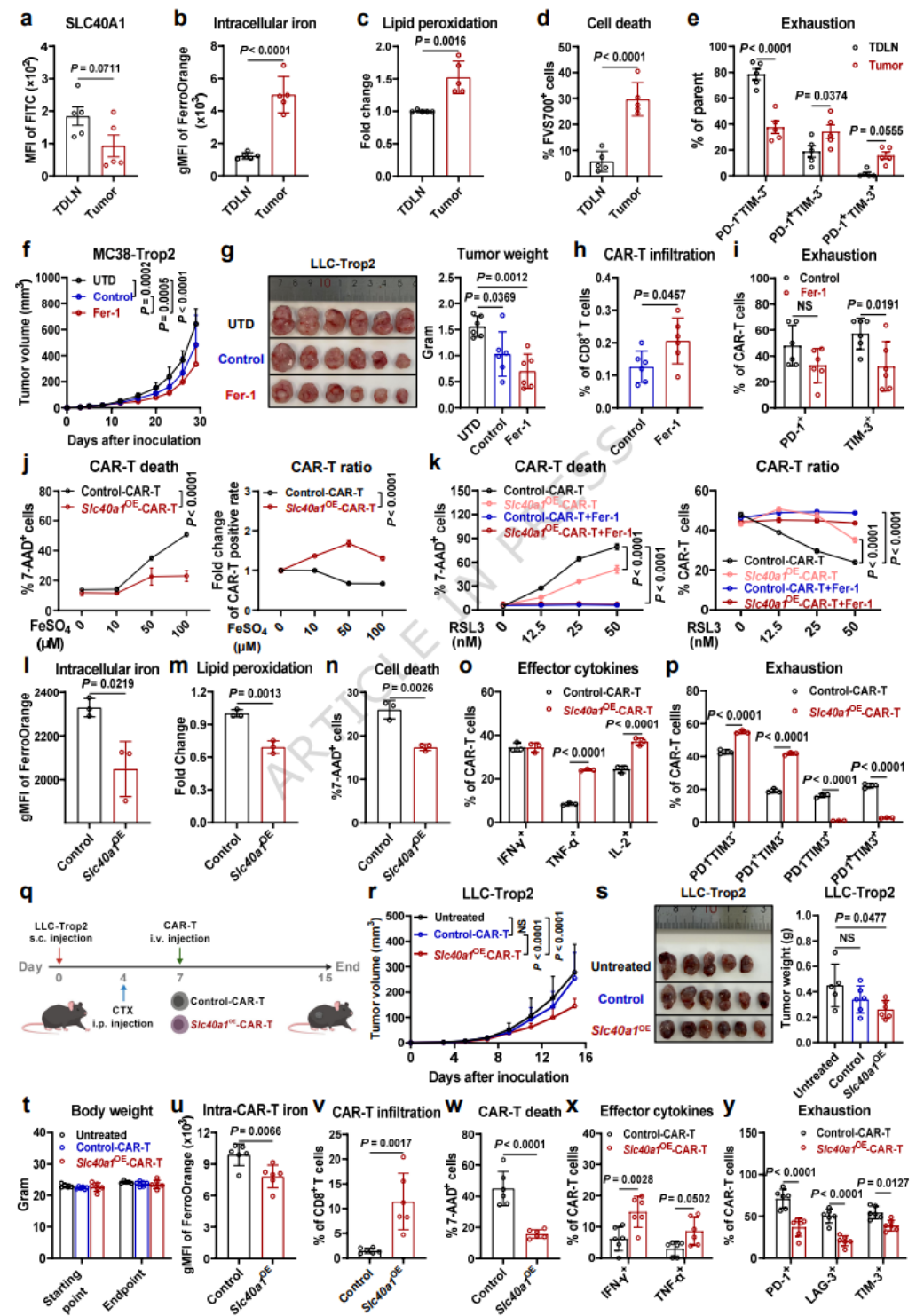
- 临床样本:结直肠癌、肺癌患者外周血缺铁,TIF与胸水铁显著富集;小鼠模型呈现一致的外周缺铁、TME铁过载表型。
- 肿瘤坏死区铁沉积更高,CD8+T细胞浸润减少、胞内铁升高;空间转录组显示坏死区铁代谢基因富集、CD8+T细胞浸润降低。
- 生理状态下淋巴液铁远低于血液,TME铁远超正常组织;肿瘤浸润CD8+T细胞胞内铁随耗竭程度逐步升高。
(二)铁过载通过铁死亡驱动CD8+T细胞功能损伤
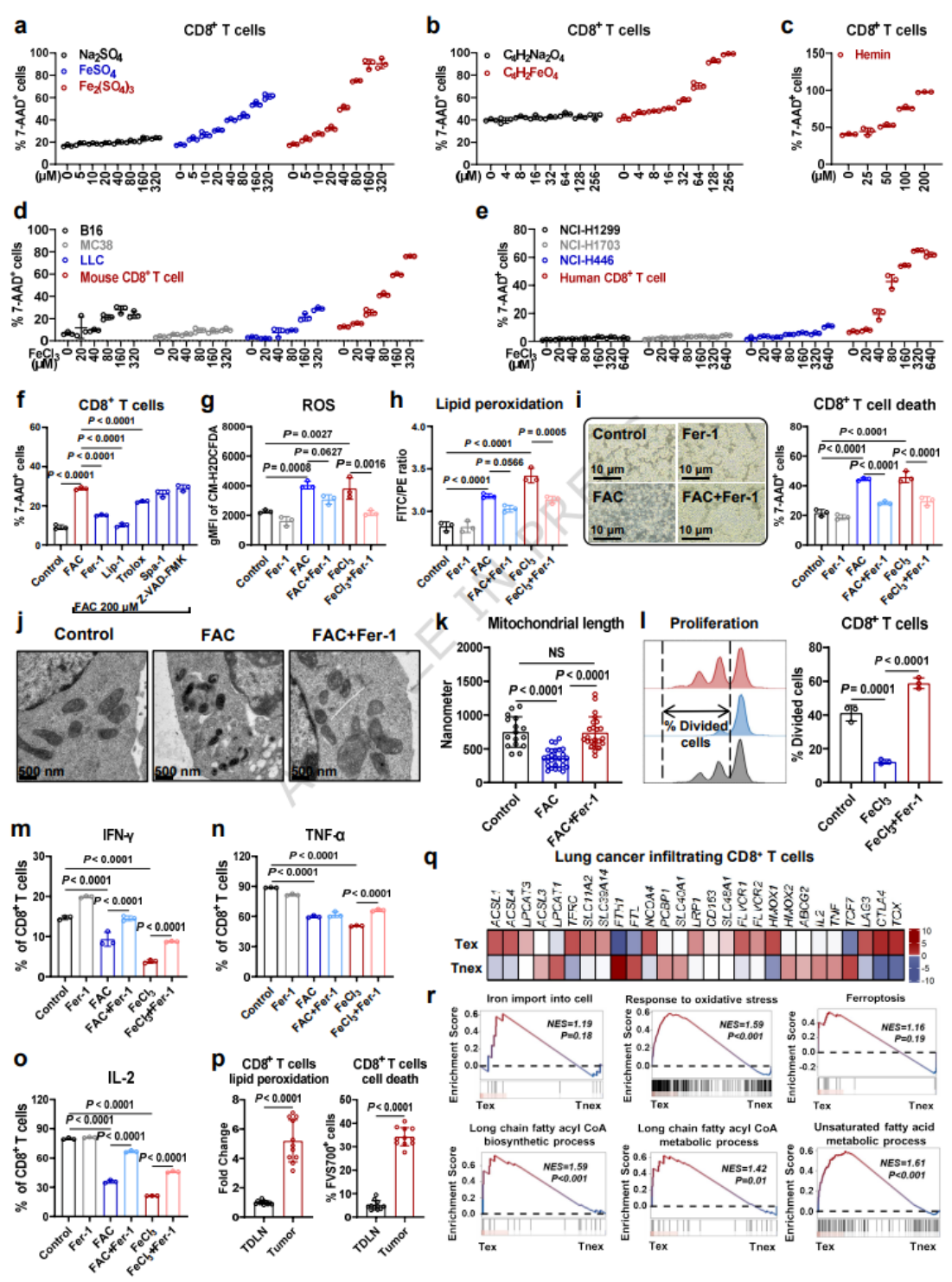
- 超生理浓度铁剂量依赖性诱导CD8+T细胞死亡,肿瘤细胞铁耐受显著高于CD8+T细胞。
- 铁死亡抑制剂可逆转铁诱导的T细胞死亡,降低ROS与脂质过氧化,挽救线粒体损伤。
- 铁过载抑制T细胞增殖,降低IFN-γ/TNF-α/IL-2分泌;体内肿瘤浸润CD8+T细胞存在显著脂质过氧化与铁死亡特征。
(三)SLC40A1下调是CD8+T细胞铁蓄积与铁死亡的核心原因
- CD8+T细胞激活后铁摄入与储存基因上调,SLC40A1蛋白升高但mRNA下降;TCR激活增强SLC40A1翻译效率。
- 慢性TCR刺激抑制SLC40A1转录与翻译,导致胞内铁过载;铁死亡抑制剂可逆转慢性TCR+铁过载导致的T细胞功能损伤。
- 人/小鼠肿瘤浸润CD8+T细胞SLC40A1显著下调;回补SLC40A1可降低铁蓄积、抑制铁死亡、恢复效应功能。

(四)肿瘤来源铁调素降解SLC40A1,加剧CD8+T细胞铁死亡
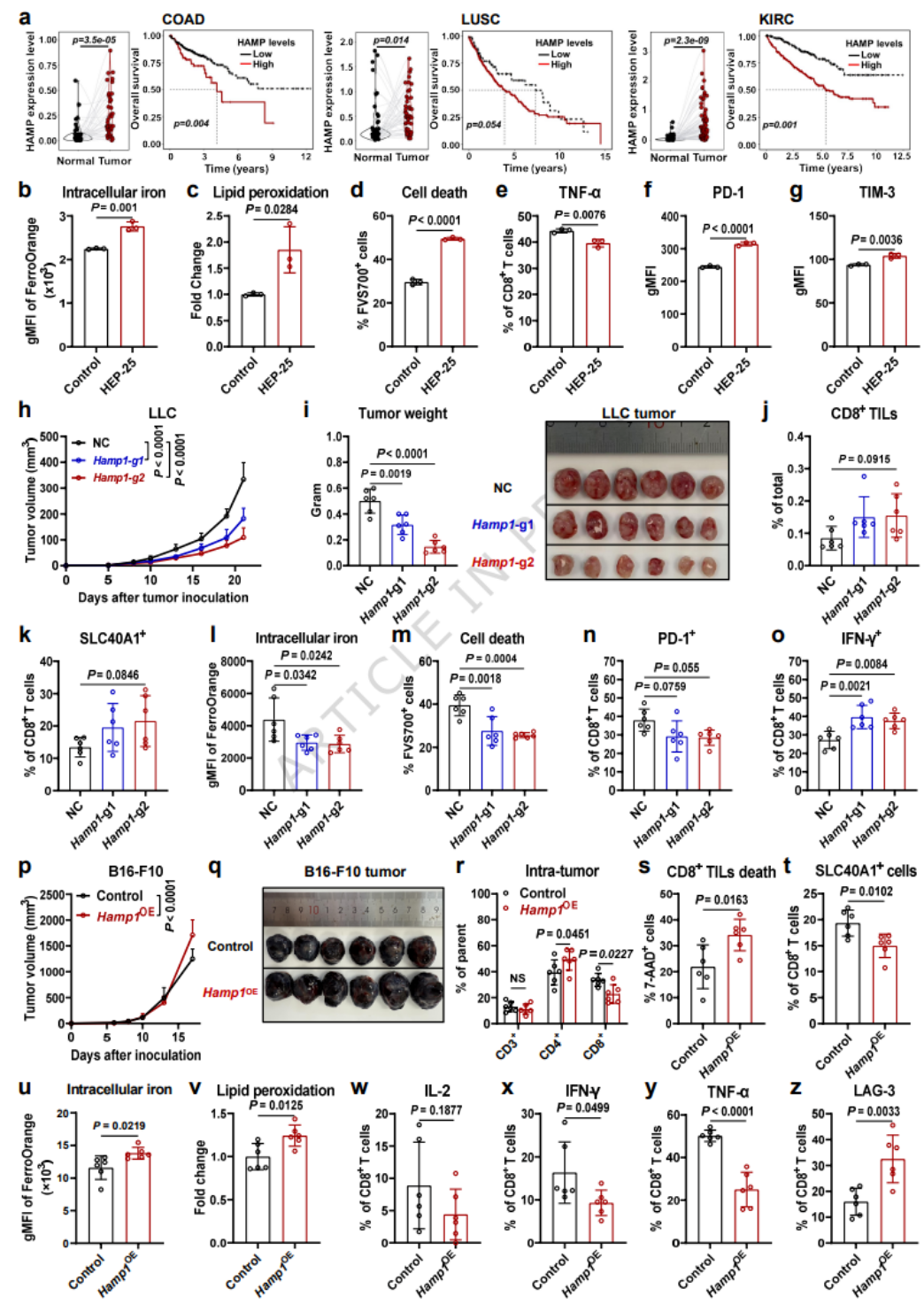
- 多种肿瘤高表达HAMP(铁调素),与患者不良预后相关。
- 铁调素处理降低SLC40A1,升高胞内铁、脂质过氧化与耗竭;肿瘤铁调素敲除减慢肿瘤生长,升高CD8+T细胞SLC40A1并增强功能。
- 铁调素过表达加速肿瘤生长,降低CD8+T细胞浸润与SLC40A1,加剧铁死亡。
(五)铁调控重塑CD8+T细胞抗肿瘤免疫
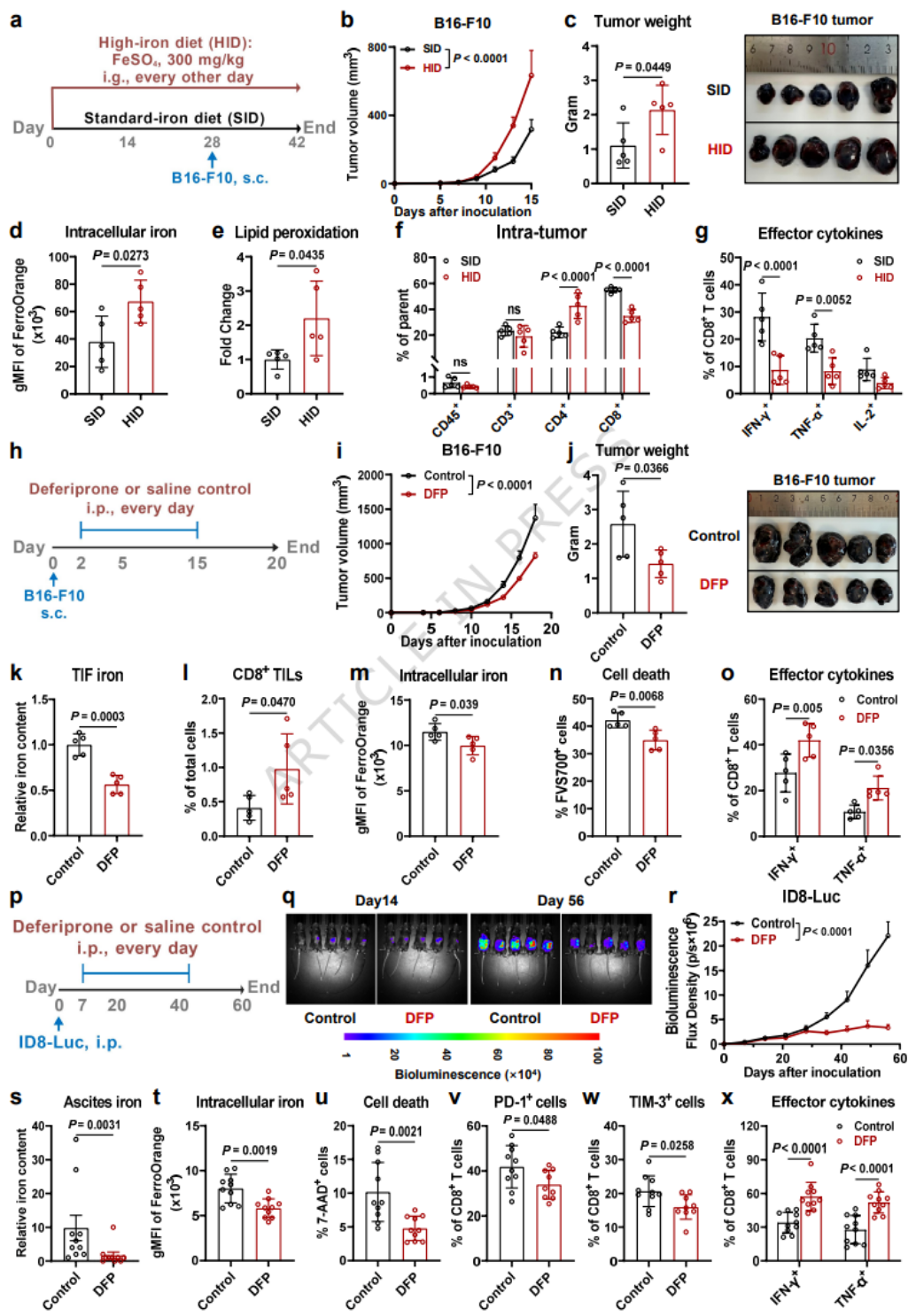
- 高铁饮食升高TME铁,加剧CD8+T细胞铁死亡与耗竭,促进肿瘤生长。
- 铁螯合剂DFP降低TME铁,减少CD8+T细胞铁死亡,恢复杀伤功能并抑制肿瘤;卵巢癌模型验证一致结果。
(六)SLC40A1过表达CAR-T抵抗铁死亡,增强实体瘤疗效
- 肿瘤浸润CAR-T细胞SLC40A1下调,呈现铁死亡与耗竭表型;铁死亡抑制剂预处理增强CAR-T在体抗肿瘤效果。
- SLC40A1过表达使CAR-T抵抗铁与铁死亡诱导剂杀伤,胞内铁降低、脂质过氧化减少、细胞因子升高、耗竭降低。
- 小鼠模型证实SLC40A1过表达CAR-T浸润更好、存活更强、抑瘤更显著。
四、讨论
本研究首次系统揭示TME铁过载诱导CD8+T细胞铁死亡与功能耗竭的全新免疫抑制机制,核心通路为:肿瘤坏死→TME铁过载→慢性TCR激活+肿瘤铁调素→SLC40A1下调→胞内铁蓄积→铁死亡→T细胞耗竭。 该发现解释了肿瘤患者全身缺铁而局部铁富集的矛盾现象,为临床补铁争议提供机制答案。SLC40A1作为关键调控节点,其过表达可直接赋予CAR-T抗铁死亡能力,突破实体瘤免疫抑制微环境瓶颈,为新一代工程化免疫细胞治疗提供靶点与策略。 未来可进一步探索铁螯合剂与免疫治疗的联合方案,优化SLC40A1过表达CAR-T的临床转化,为实体瘤治疗提供新方向。
五、结论
1.肿瘤微环境铁过载是CD8+T细胞铁死亡与功能耗竭的重要诱因。
2.慢性TCR信号与肿瘤铁调素双重抑制SLC40A1是核心分子机制。
3.恢复SLC40A1表达或铁螯合可逆转CD8+T细胞功能损伤。
4.SLC40A1过表达CAR-T可抵抗铁过载,显著提升实体瘤治疗效果。








 沪公网安备31011502400759号
沪公网安备31011502400759号
 营业执照(三证合一)
营业执照(三证合一)




