













巨噬细胞作为先天免疫与适应性免疫的关键枢纽,在机体稳态维持与免疫应答中发挥核心作用。在肿瘤微环境中,巨噬细胞分化为具有高度可塑性的TAMs,其极化方向直接调控肿瘤免疫状态。近年研究证实,巨噬细胞极化依赖代谢重编程的支撑:M1型巨噬细胞以有氧糖酵解为主导代谢模式,M2型巨噬细胞则依赖氧化磷酸化(OXPHOS)与脂肪酸氧化(FAO)。这种代谢-极化的协同调控不仅是巨噬细胞功能特化的基础,也是肿瘤微环境免疫抑制形成的关键机制。深入解析二者的交互作用分子机制,是当前肿瘤免疫代谢领域的核心科研方向,可为开发新型调控策略提供靶点支撑。
一、肿瘤微环境的代谢特征及免疫细胞调控机制
肿瘤微环境的代谢重构是肿瘤进展的典型特征,其代谢产物与营养竞争直接调控免疫细胞功能,形成独特的免疫代谢网络。
(一)代谢因素对免疫细胞的选择性调控
肿瘤微环境中多种代谢改变通过特异性机制调控不同免疫细胞功能:
CD8⁺T细胞:肿瘤细胞糖酵解产生的乳酸大量积累,可抑制CD8⁺T细胞的代谢活性,导致其增殖停滞及IFN-γ等细胞因子分泌减少;而阻断肿瘤细胞代谢重编程可恢复CD8⁺T细胞的代谢活性与抗肿瘤功能,这一发现为解析肿瘤免疫逃逸机制提供了重要线索。
NKT细胞:肝癌微环境中胆汁酸可通过特异性受体促进NKT细胞浸润,增强其细胞毒性,揭示了代谢产物调控特异性免疫细胞募集的新机制。
调节性T细胞(Treg):肿瘤微环境的低糖状态可通过激活STAT3信号通路诱导Treg细胞分化,其免疫抑制功能进一步加剧肿瘤免疫耐受,为探索Treg细胞代谢依赖的分化机制提供了研究切入点。
髓系来源抑制性细胞(MDSCs):肿瘤细胞糖酵解增强可上调G-CSF/GM-CSF的分泌,通过趋化信号招募MDSCs至肿瘤局部,后者通过产生活性氧与精氨酸酶抑制T细胞功能,形成“糖酵解-MDSCs-免疫抑制”的调控轴。
(二)代谢重编程与免疫检查点分子的关联
免疫检查点分子(PD-1/PD-L1)的表达调控与代谢状态密切相关:肥胖相关的高脂微环境可上调肿瘤细胞与免疫细胞表面PD-1/PD-L1的表达水平,但黑色素瘤、非小细胞肺癌等肿瘤中,肥胖患者接受PD-1/PD-L1抑制剂治疗时疗效更优。这一矛盾现象提示,代谢状态可能通过调控免疫细胞的代谢适应性影响免疫检查点疗法的应答效率,为探索“代谢-免疫检查点”交互调控机制提供了重要科研方向。
二、巨噬细胞极化与代谢重编程的双向交互机制
巨噬细胞极化是代谢重编程的功能体现,而代谢通路的动态调控又可反向塑造极化表型,形成精密的分子调控网络。
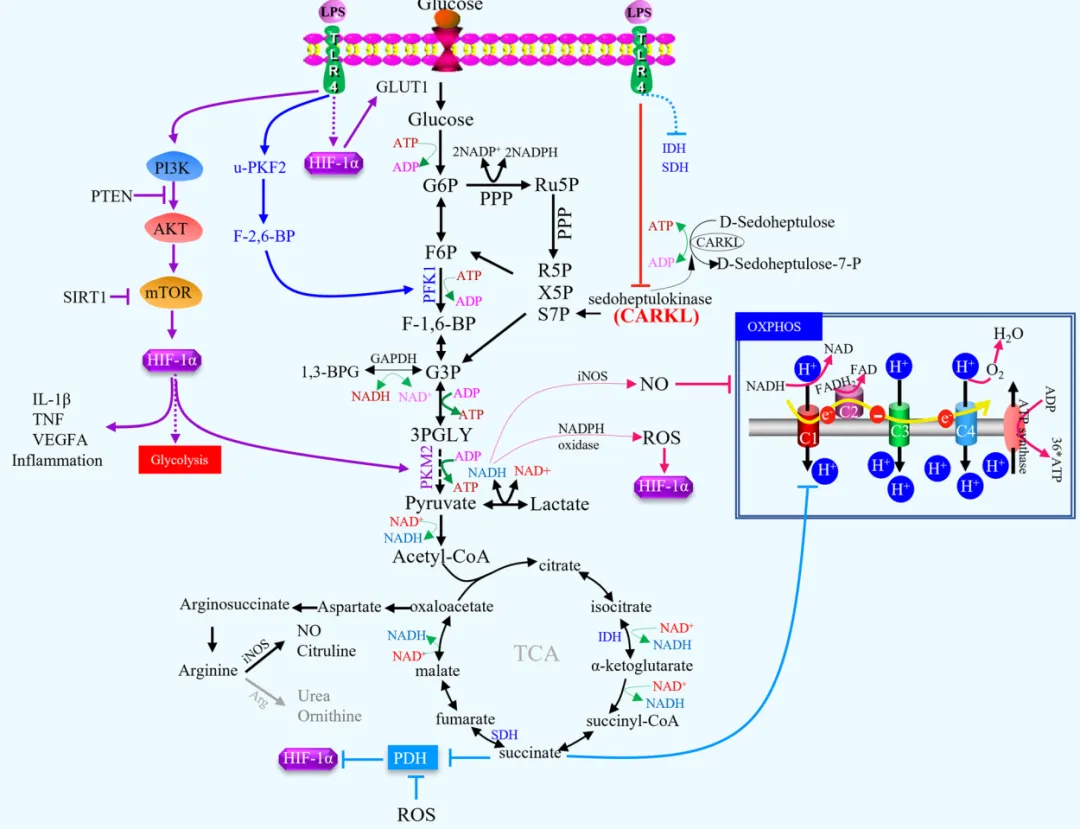
(一)M1型巨噬细胞的代谢特征与分子调控
M1型巨噬细胞的促炎功能依赖特异性代谢重编程:
代谢表型:有氧糖酵解显著增强(Warburg效应),OXPHOS受抑;磷酸戊糖途径(PPP)活跃,NADPH生成增加以支撑ROS产生;诱导型一氧化氮合酶(iNOS)高表达,精氨酸酶1(Arg1)活性低;COX-2高表达、COX-1低表达,谷胱甘肽水平升高以维持氧化还原平衡。
核心调控机制:HIF-1α作为关键转录因子,激活后可上调糖酵解关键酶(PFKFB3、GLUT1)与IL-1β等促炎因子表达;PFKFB3作为糖酵解限速酶,其上调可显著增强糖酵解通量,是M1极化的标志性代谢事件;SHPK(CARKL)表达下调可解除对NF-κB信号的抑制,促进M1极化;琥珀酸累积可通过稳定HIF-1α延长M1型炎症反应,而NADPH/ROS比值升高可激活NF-κB通路,促进PD-L1表达。
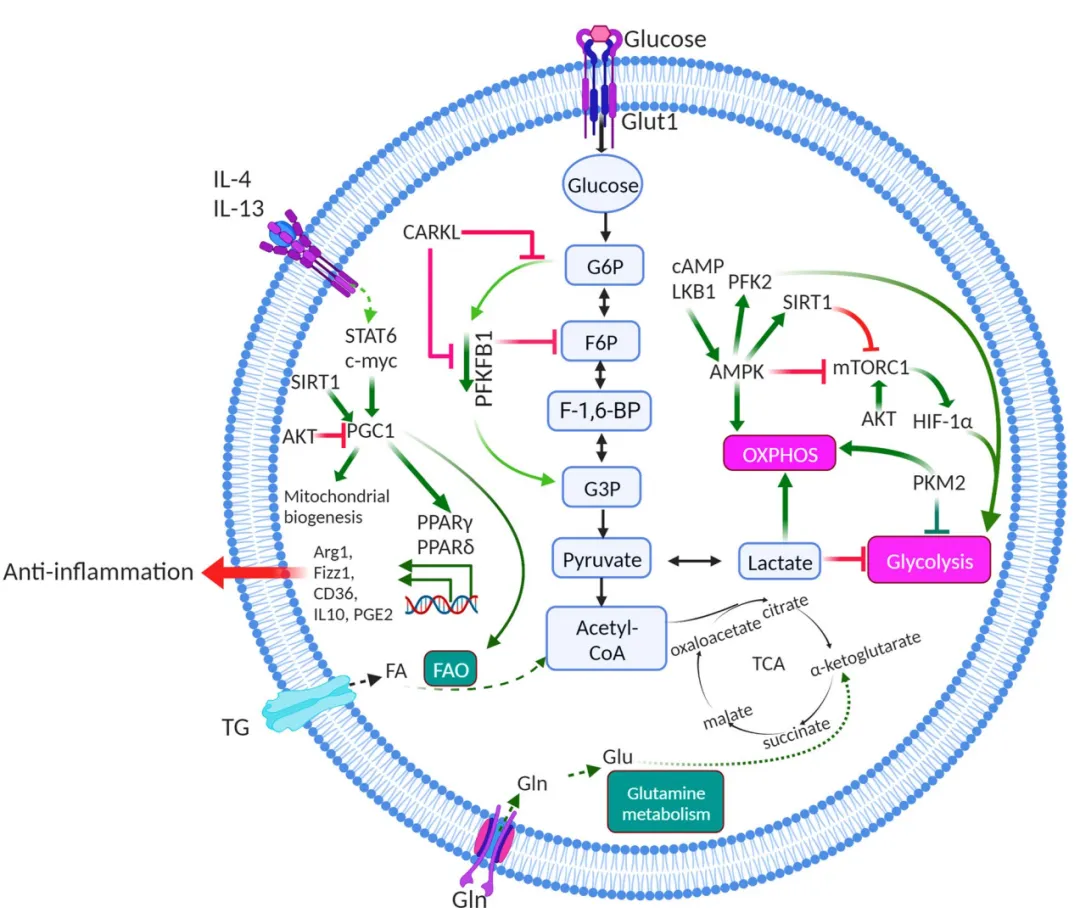
(二)M2型巨噬细胞的代谢特征与分子调控
M2型巨噬细胞的抗炎表型依赖代谢模式的特异性转换:
1. 核心代谢表型
- 有氧糖酵解显著增强(Warburg效应),氧化磷酸化(OXPHOS)受抑制;
- 磷酸戊糖途径(PPP)活跃,NADPH生成增加,为活性氧(ROS)产生提供支持;
- 诱导型一氧化氮合酶(iNOS)高表达,精氨酸酶1(Arg1)活性低;
- 环氧化酶2(COX-2)高表达、COX-1低表达,谷胱甘肽水平升高,维持氧化还原平衡;
2. 关键调控机制
- HIF-1α(核心转录因子):激活后上调糖酵解关键酶(PFKFB3、GLUT1)及IL-1β等促炎因子表达;
- PFKFB3(糖酵解限速酶):表达上调增强糖酵解通量,是M1极化的标志性代谢事件;
- SHPK(CARKL):表达下调解除对NF-κB信号的抑制,促进M1极化;
- 琥珀酸:累积可稳定HIF-1α,延长M1型炎症反应;
- NADPH/ROS比值:升高激活NF-κB通路,促进PD-L1表达;
(三)代谢干预对巨噬细胞极化的调控潜力
靶向关键代谢节点可实现巨噬细胞极化状态的人工调控:抑制糖酵解抑制剂(2-DG)可阻断M1型巨噬细胞的促炎功能;FAO抑制剂(Etomoxir)可显著抑制M2极化,逆转其抗炎表型;而靶向SHPK、PFKFB3、HIF-1α等核心调控因子,可实现TAMs极化状态的双向转换,这为后续科研中开发极化调控工具提供了重要依据。
三、关键代谢通路对TAMs极化的特异性调控
(一)缺氧微环境的调控机制
TAMs常聚集于肿瘤缺氧区域,慢性缺氧通过HIF-1α依赖机制调控其功能:缺氧可促进葡萄糖进入磷酸戊糖途径,增加NADPH生成以维持氧化还原平衡,同时促进吞噬体成熟,增强TAMs对凋亡细胞的吞噬功能;HIF-1α可通过激活PKM2、PFKFB3增强糖酵解,上调iNOS、IL-1β等炎症因子表达,通过FSTL1-PKM2磷酸化通路促进M1极化,过度表达时可诱导上皮-间质转化(EMT)促进肿瘤转移,还可通过抑制FoxO1下调MHC-II表达,削弱抗原呈递功能。
(二)脂质代谢的调控网络
脂质代谢通过多条通路调控TAMs极化,是免疫代谢领域的研究热点:
- 脂肪酸氧化(FAO):M2型巨噬细胞依赖FAO维持线粒体代谢与抗炎功能,而M1型巨噬细胞中FAO受抑;
- 胆固醇代谢:胆固醇在M1型巨噬细胞中积聚可诱导炎症反应,而M2型巨噬细胞通过增强胆固醇外排促进极化;
- 脂质摄取:M2型巨噬细胞高表达CD36分子,通过增强脂质摄取为FAO提供底物;
- 花生四烯酸代谢:M1型巨噬细胞通过COX-2产生促炎介质PGE2,M2型则通过COX-1/ALOX15产生抗炎介质resolvin,调控炎症与修复平衡;
- 甘油三酯代谢:M2型巨噬细胞中MGLL活性升高,促进脂质积聚,其缺失可通过CB2/TLR4通路促进M2激活。
PPAR家族(α/γ/δ)与PGC-1β是脂质代谢调控极化的核心分子:PPARγ与PGC-1β协同诱导Arg1表达,推动M2极化,其激动剂可调控TAMs代谢表型;PPARδ与STAT6协同促进M2极化,调控脂肪酸代谢基因表达。
(三)氨基酸代谢的调控作用
氨基酸代谢通过特异性通路调控巨噬细胞极化方向:
- L-精氨酸代谢:通过iNOS(M1型)与Arg1(M2型)两条分支代谢决定极化方向,是巨噬细胞功能特化的关键节点;
- 支链氨基酸酮酸(BCKAs):胶质瘤细胞分泌的BCKAs被TAMs摄取后,可抑制其吞噬功能,阻碍M1极化;
- 丝氨酸代谢:为LPS诱导IL-1β mRNA表达所必需,是M1型巨噬细胞促炎反应的代谢基础;
关键分子:
- SLC7A5(亮氨酸转运体)通过激活mTORC1促进M1极化;
- ODC(多胺合成限速酶)抑制M1激活;
- ARG2(线粒体精氨酸酶)可诱导M1极化并促进促炎因子分泌。
四、科研展望
巨噬细胞极化与代谢重编程的交互作用是肿瘤免疫代谢领域的核心科学问题,当前研究已揭示了糖酵解、OXPHOS、脂质代谢等关键通路的调控作用,但仍存在诸多科研空白:不同肿瘤类型中TAMs代谢表型的异质性机制、代谢产物调控极化的特异性受体识别、表观遗传修饰与代谢重编程的协同调控网络等,均需进一步深入探索。
未来科研应聚焦核心代谢酶(如PFKFB3、Arg1)与转录因子(如HIF-1α、PPARγ)的靶向调控,开发高特异性的代谢干预工具,解析代谢-极化轴在肿瘤进展中的动态调控规律,为后续靶向TAMs代谢的科研转化提供理论支撑。








 沪公网安备31011502400759号
沪公网安备31011502400759号
 营业执照(三证合一)
营业执照(三证合一)




