







神经系统疾病的病理进程复杂多样,从急性损伤(如脑卒中)到慢性退行性病变(如阿尔茨海默病、帕金森病),尽管病因迥异,但均伴随神经炎症的动态参与。神经炎症具有双重生物学特性:早期通过清除病原体、降解损伤组织碎片发挥防御修复功能,而调控失衡时则通过蛋白酶释放、氧化应激、细胞因子风暴等机制演变为病理性炎症,加剧神经元损伤,并形成"神经炎症-神经退行性变"的恶性循环。 本文基于2025年6月施福东教授与V. Wee Yong教授在《Science》发表的综述成果,聚焦科研端核心问题,系统解析神经炎症的启动机制、核心调控网络,深入探讨四大代表性神经系统疾病中神经炎症的病理特征差异与共性规律,重点阐述炎症调控关键节点的科研突破及新型靶点开发方向,为神经系统疾病的免疫调控研究提供理论框架与科研思路。

一、神经炎症的核心病理机制与调控网络
神经炎症的发生发展是固有免疫与适应性免疫协同作用的结果,其核心构成包括CNS固有细胞激活、免疫细胞浸润及驱动分子网络三大关键组分,三者相互作用形成复杂的调控环路。
(一)CNS固有细胞的激活机制
小胶质细胞作为中枢神经系统常驻免疫细胞,是神经炎症的"启动者"。其激活依赖于损伤相关分子模式(DAMPs)和病原体相关分子模式(PAMPs)的识别:当CNS发生损伤或出现异常蛋白聚集时,小胶质细胞通过表面受体识别上述信号,快速启动活化程序,表现为形态改变、功能重塑及细胞因子分泌。 星形胶质细胞作为CNS内数量最多的胶质细胞,通过与小胶质细胞的交叉对话参与炎症调控:活化的小胶质细胞分泌的促炎因子可诱导星形胶质细胞激活,而星形胶质细胞进一步分泌趋化因子与促炎因子,形成炎症放大环路,加剧局部炎症反应。二者的功能协同与失衡是神经炎症从保护向损伤转换的关键环节。
(二)免疫细胞浸润的分子机制与调控
外周免疫细胞跨越血脑屏障(BBB)或软脑膜屏障进入CNS是神经炎症加剧的核心事件,其分子机制涉及多层面调控:
1.屏障通透性调控:病理状态下,DAMPs、促炎因子等信号可诱导BBB内皮细胞紧密连接蛋白表达下调,增加屏障通透性,为外周免疫细胞浸润提供结构基础;
2.细胞迁移信号:趋化因子(如CCL2、CXCL10)及其受体的相互作用为免疫细胞迁移提供定向信号,其中T细胞、单核细胞来源巨噬细胞(MdM)、中性粒细胞是主要浸润细胞类型;
3.细胞间互作:浸润的T细胞与小胶质细胞的直接互作是病理性炎症放大的关键节点,通过抗原呈递、共刺激分子激活等机制,推动小胶质细胞向致损伤表型转换,同时促进T细胞增殖与细胞因子分泌,形成级联放大效应。
(三)炎症驱动的分子网络调控
神经炎症的持续与放大依赖于复杂的分子网络,核心驱动分子包括:
1.始动信号分子:DAMPs(如髓鞘碎片、β淀粉样蛋白、α-突触核蛋白等错误折叠蛋白)作为炎症启动的"危险信号",直接激活固有免疫细胞; ‘’
2.免疫调控分子:补体成分C1q通过介导星形胶质细胞神经毒性表型转换参与炎症放大;骨桥蛋白(OPN)则通过调控免疫细胞迁移与活化,促进炎症持续;
3.效应分子:活性氧(ROS)、促炎细胞因子(TNF-α、IL-1α)等作为下游效应分子,直接介导神经元损伤、轴索退变及突触功能异常,是神经炎症病理损伤的最终执行者。
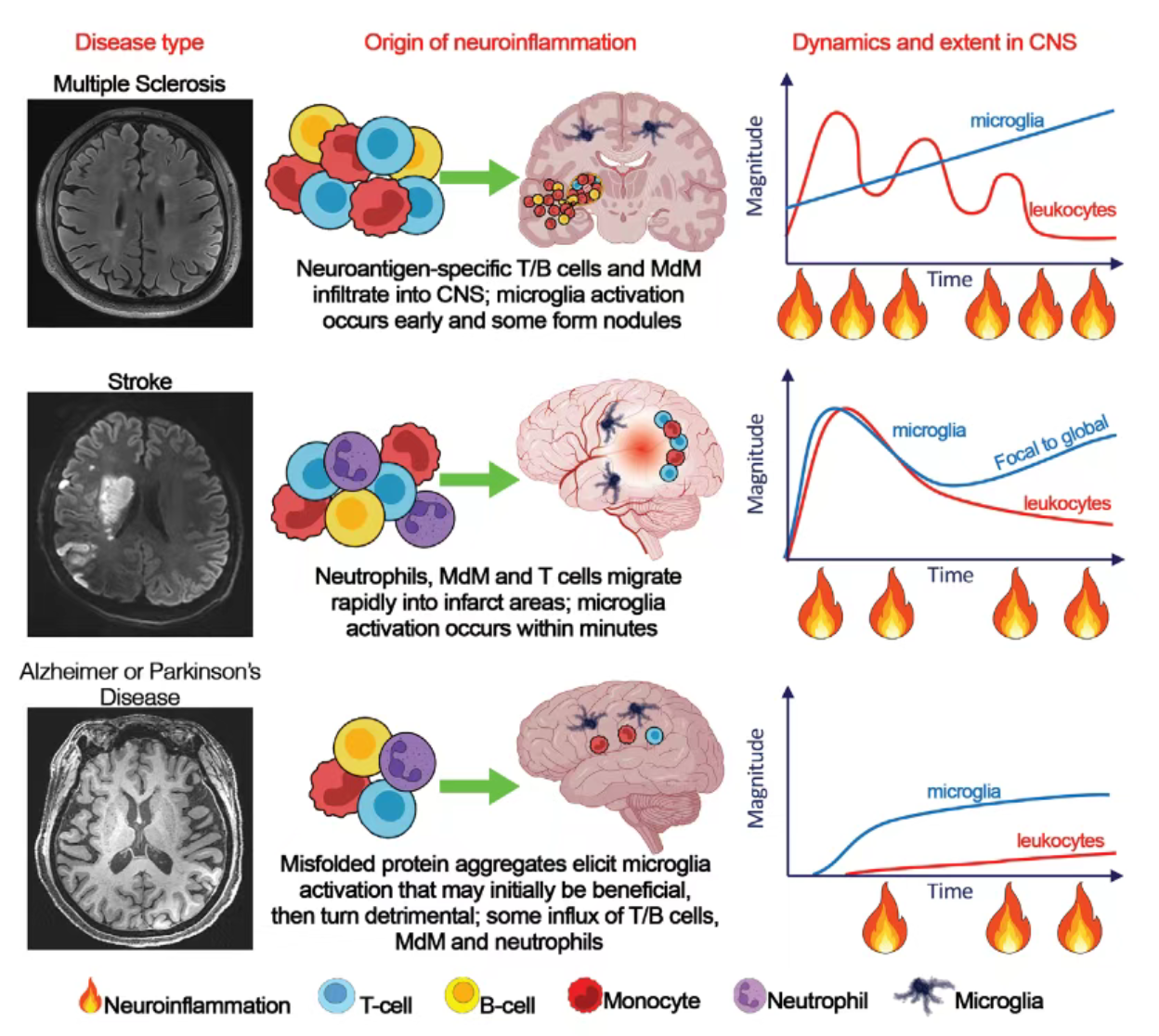
图1:神经炎症在MS、卒中、AD和PD中的基础
二、四大神经系统疾病的神经炎症科研特征解析
(一)多发性硬化:自身免疫驱动的炎症病理模型
多发性硬化(MS)作为免疫介导型疾病,其神经炎症的科研核心特征为"外周起源-中枢放大":
1.启动机制:分子模拟机制是CNS反应性T细胞激活的关键,激活的CD8⁺ T细胞(病灶中占比高于CD4⁺ T细胞)是早期浸润的核心效应细胞,其克隆扩增与持续性浸润是疾病进展的重要标志;
2.免疫细胞构成:小胶质细胞与MdM占病灶免疫细胞的80%以上,其密度与轴索损伤程度呈正相关,且从疾病初期即呈现致损伤表型,其功能调控是科研关注的核心靶点;
3.特殊病理特征:脑膜与血管周围的B细胞聚集形成特异性免疫微环境,通过抗原呈递、抗体分泌等机制加剧皮质损伤,而慢性活动性病灶(CALs)的小胶质细胞持续激活是疾病慢性进展的关键驱动因素,为疾病机制研究提供了重要病理模型。
(二)脑卒中:急性损伤触发的炎症时序模型
脑卒中的神经炎症以"急性快速应答"为核心科研特征,其炎症动态遵循明确的时序规律:
1.时序激活模式:"分钟级小胶质细胞激活→小时级中性粒细胞浸润→天级单核细胞与T细胞募集",不同阶段的免疫细胞构成与功能差异为炎症分期调控研究提供了天然模型;
2.功能转换机制:早期小胶质细胞激活以清除坏死组织、促进组织修复为主要功能,而后期通过ROS与蛋白酶释放加剧BBB破坏,其功能转换的分子开关(如转录因子NF-κB的激活阈值)是科研关键问题;
3.扩散机制:炎症反应从梗死灶局部向全脑扩散的分子路径(如神经环路传播、体液信号介导),及其与卒中后远期并发症(痴呆、抑郁)的关联,是当前科研的重要探索方向。
(三)阿尔茨海默病:蛋白聚集驱动的慢性炎症模型
阿尔茨海默病(AD)的神经炎症以β淀粉样蛋白(Aβ)沉积和tau过度磷酸化为始动因素,科研核心聚焦"保护-损伤"双相转换:
1.小胶质细胞功能调控:早期小胶质细胞通过吞噬作用清除Aβ聚集体,该功能受TREM2、APOE等基因调控,其中TREM2突变可显著降低小胶质细胞吞噬能力,增加AD发病风险,成为基因调控机制研究的核心靶点;
2.细胞互作机制:小胶质细胞与星形胶质细胞的交叉对话(C1q介导)是疾病进展的关键,C1q可诱导星形胶质细胞表达神经毒性分子,同时抑制小胶质细胞吞噬功能,其分子机制是炎症放大研究的重点;
3.外周免疫参与:CD8⁺效应记忆T细胞的低密度浸润及其与中枢免疫细胞的互作模式,虽浸润密度低于MS,但对炎症慢性化的调控作用仍为科研热点。
(四)帕金森病:α-突触核蛋白相关的炎症病理机制
帕金森病(PD)的神经炎症科研核心围绕α-突触核蛋白(α-syn)聚集展开:
1.清除机制:小胶质细胞通过隧道纳米管介导α-syn聚集体清除,而LRRK2突变可破坏该清除机制,导致α-syn异常蓄积,其分子机制为PD炎症启动研究提供了关键线索;
2.免疫细胞互作:CD4⁺/CD8⁺ T细胞在路易小体周围聚集,与小胶质细胞的互作可促进小胶质细胞过度激活,其抗原识别特异性与激活信号通路是科研核心问题;
3.星形胶质细胞调控:通过NF-κB、p38 MAPK通路激活的星形胶质细胞,其促炎因子与ROS分泌的调控机制,及其对黑质多巴胺能神经元的直接损伤作用,是PD炎症损伤机制的研究重点。
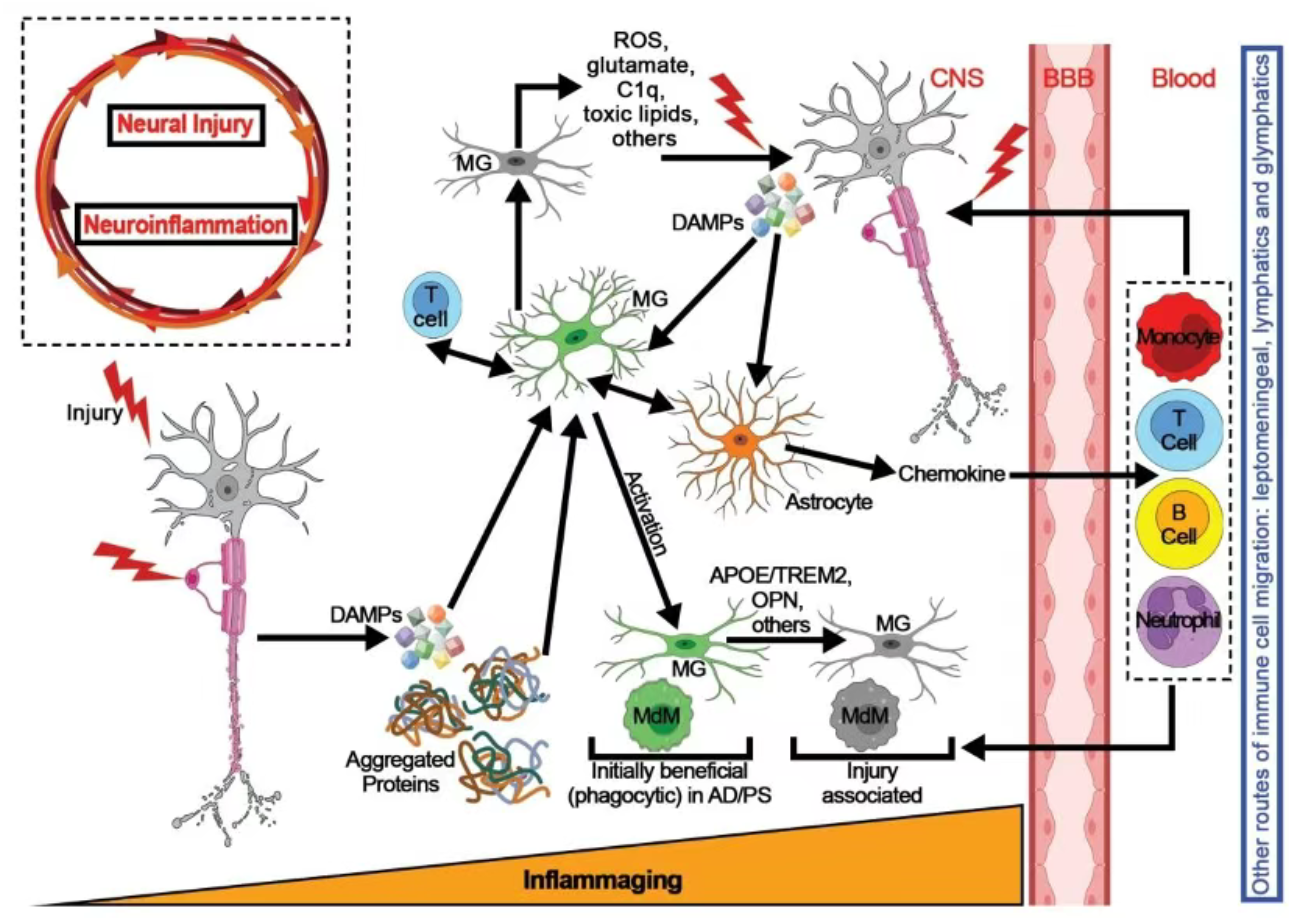
图2. 神经炎症与神经损伤通过前馈循环相互促进
(五)四大疾病神经炎症的共性与差异
1. 共性规律
核心细胞事件:小胶质细胞激活是所有疾病神经炎症的起始与核心事件,其功能转换是炎症病理损伤的关键;
调控核心节点:T细胞-小胶质细胞/星形胶质细胞互作是炎症放大的保守机制,在不同疾病中均发挥关键作用;
病理演变趋势:神经炎症均呈现从"外周主导"向"中枢局限化"的演变,且均形成"神经炎症-神经退行性变"恶性循环,为跨疾病研究提供了理论基础。
2. 差异焦点
启动机制差异:MS为自身免疫驱动,卒中为急性损伤触发,AD/PD为错误折叠蛋白诱导,不同启动机制为针对性靶点开发提供了方向;
细胞功能差异:小胶质细胞在AD/PD中呈现"保护-损伤"双相转换,而在MS中起始即表现为致损伤表型,其分子调控网络的差异是科研核心探索点;
免疫细胞参与度:MS中外周免疫细胞(T/B细胞、MdM)浸润显著且持续,AD/PD中浸润密度低但功能关键,这种差异为疾病分型研究提供了病理依据。优化转化研究体系,有望实现神经炎症从机制研究向精准干预的跨越,为神经系统疾病的科研突破与临床转化提供新的路径。
三、总结
神经炎症作为跨越多种神经系统疾病的核心病理机制,其科研核心聚焦于固有免疫与适应性免疫的协同调控、功能转换及病理损伤机制。小胶质细胞激活、免疫细胞浸润、分子网络调控构成了神经炎症的三大核心科研方向,而不同神经系统疾病的炎症特征差异为针对性研究提供了依据,共性规律则为跨疾病治疗策略开发奠定了基础。
当前,随着单细胞测序、空间转录组等技术的应用,神经炎症的调控机制研究已进入精准化、分子化阶段,新型靶点的不断挖掘与验证为科研转化提供了重要支撑。未来LabEx 将以标准化实验操作、严谨的数据分析流程与全链条技术服务,助力科研人员完成神经炎症从机制研究到精准干预的跨越,为神经系统疾病的科研突破与临床转化开辟更高效的全新路径。
LabEx多款现货炎症因子Panel,欢迎咨询~
| 官网货号 | Panel | 技术平台 | 检测指标 |
| LXMH10-1 | 人炎症10因子Panel | MSD | IFN-γ,IL-1β,IL-2,IL-4,IL-6,IL-8,IL-10,IL-12p70,IL-13,TNF-α |
| LXLBH10-1 | 人炎症10因子Panel | Luminex | IL-1 β/IL-1F2,IL-2,IL-4,IL-6 ,IL-8/CXCL8,IL-10,IL-12 p70,IL-13,TNF-α,IFN-γ |
| LXMM10-1 | 小鼠炎症10因子Panel | MSD | IFN-γ,IL-1β,IL-2,IL-4,IL-5,IL-6,IL-10,IL-12p70,KC/GRO,TNF-α |
| LXLBM10-1 | 小鼠炎症10因子Panel | Luminex | IL-1 β/IL-1F2,IL-2,IL-4,IL-5,IL-6 ,IL-10,IL-12p70,CXCL1/GRO/α/KC/CINC-1,IFN-γ,TNF-α |
| LXLBR10-1 | 大鼠炎症10因子Panel | Luminex | IL-1 β/IL-1F2,IL-2,IL-4,IL-5,IL-6 ,IL-10,IL-12p70,CXCL1/GRO/α/KC/CINC-1,IFN-γ,TNF-α |
| LXLBM31-1 | 小鼠趋化因子-31因子Panel | Luminex | BCA-1/CXCL13,CTACK/CCL27,ENA-78/CXCL5,Eotaxin/CCL11,Eotaxin-2/CCL24,Fractalkine/CX3CL1,GM-CSF,I-309/CCL1,IFN-γ,IL-1β,IL-2,IL-4,IL-6,IL-10,IL-16,IP-10/CXCL10,I-TAC/CXCL11,KC/CXCL1,MCP-1/CCL2,MCP-3/CCL7,MCP-5/CCL12,MDC/CCL22,MIP-1α/CCL3,MIP-1β/CCL4,MIP-3α/CCL20,MIP-3β/CCL19,RANTES/CCL5,SCYB16/CXCL16,SDF-1α/CXCL12,TARC/CCL17,TNF-α |
| LXLRH46-1 | 人细胞因子-46因子Panel | Luminex | CCL2/JE/MCP-1,CCL3/MIP-1 alpha,CCL4/MIP-1 beta,CCL5/RANTES,CCL11/Eotaxin,CCL19/MIP-3 beta,CCL20/MIP-3 alpha,CD40 Ligand/TNFSF5,CXCL1/GRO alpha/KC/CINC-1,CXCL2/GRO beta/MIP-2/CINC-3,CXCL10/IP-10/CRG-2,EGF,FGF basic/FGF2/bFGF,Flt-3 Ligand/FLT3L,G-CSF,GM-CSF,Granzyme B,IFN-alpha 2/IFNA2,IFN-beta,IFN-gamma,IL-1 alpha/IL-1F1,IL-1 beta/IL-1F2,IL-1ra/IL-1F3,IL-2,IL-3,IL-4,IL-5,IL-6,IL-7,IL-8/CXCL8,IL-9,IL-10,IL-12 p70,IL-13,IL-15,IL-17/IL-17A,IL-17E/IL-25,IL-33,Lymphotoxin-alpha/TNF-beta,PD-L1/B7-H1,PDGF-AA,PDGF-AB/BB,TGF-alpha,TNF-alpha,TRAIL/TNFSF10,VEGF |
| LXLBH48-1 | 人细胞因子-48因子Panel | Luminex | β-NGF,CTACK/CCL27,Eotaxin/CCL11,FGF-basic,G-CSF,GM-CSF,GRO-α (Gro-a/KC/CXCL1),HGF,IFN-α2,IFN-γ,IL-1α,IL-1Rα,IL-2Rα,IL-1β,IL-2,IL-3,IL-4,IL-5,IL-6,IL-7,IL-8/CXCL8,IL-9,IL-10,IL-12(p40),IL-12(p70),IL-13,IL-15,IL-16,IL-17A,IL-18,IP-10/CXCL10,LIF,M-CSF,MCP-1/CCL2,MCP-3/CCL7,MIG,MIP-1α/CCL3,MIP-1β,MIF,PDGF-BB,RANTES,SCF,SCGF-β,SDF-1α,TRAIL,TNF-α,TNF-β,VEGF-A |








 沪公网安备31011502400759号
沪公网安备31011502400759号
 营业执照(三证合一)
营业执照(三证合一)




